Вступ
За визначенням Європейського товариства кардіологів та Європейського респіраторного товариства 2015 р., під легеневою гіпертензією (ЛГ) розуміють гемодинамічний та патофізіологічний стан, що характеризується підвищенням середнього тиску в легеневій артерії (серТЛА) > 25 мм рт.ст., який виміряний під час катетеризації правих відділів серця (КПС) [1]. У 2018 році на 6-му Всесвітньому конгресі з легеневої гіпертензії змінено критерій ЛГ для величини серТЛА на > 20 мм рт.ст. [2]. З огляду на дане визначення ЛГ — це не окрема хвороба (в переважній кількості випадків), а стан, який може супроводжувати інші захворювання.
На сьогодні виділяють близько 50 хвороб, які характеризуються підвищенням тиску в ЛА. Усі вони, згідно з етіопатогенетичною класифікацією, поділяються на 5 груп [1]. Найчисленнішою є група 2: ЛГ, пов’язана із захворюванням лівих відділів серця — 80 % серед всіх ЛГ. Друге місце належить ЛГ, асоційованим із гіпоксією та захворюванням легень (група 3) — 10 %. Пацієнти груп 2 та 3, як правило, не потребують призначення специфічної терапії. Групи 1 (легенева артеріальна гіпертензія) та 4 (хронічна тромбоемболічна ЛГ) становлять менше 10 %. Це форми ЛГ, які мають схожий патогенез (ремоделювання легеневих артеріол), лікуються специфічними препаратами та які можна вважати окремими захворюваннями, незважаючи на те, що вони виникають на фоні інших хвороб (вроджені вади серця, захворювання сполучної тканини, тромбоемболія легеневих артерій, ВІЛ-інфекція, портальна гіпертензія тощо). Група 5 — це складні форми ЛГ, які мають ознаки ЛГ, що належать до груп 1–3. Ці пацієнти можуть лікуватися специфічними препаратами при певних ситуаціях.
Особливостями ЛГ, що належить до груп 2 та 3, є:
— вони зустрічаються найчастіше;
— як правило, характеризуються найгіршим прогнозом;
— на сьогодні не існує специфічного лікування, а лише рекомендації щодо лікування основного захворювання та індивідуального підходу в можливому виборі специфічної терапії.
Саме із цими групами ЛГ частіше стикаються сімейні лікарі, кардіологи, пульмонологи та кардіохірурги, але недостатні знання щодо патогенезу виникнення та рекомендацій із ведення таких пацієнтів часто призводять до необґрунтованого призначення дорогої специфічної терапії ЛГ, яка здебільшого або не змінює, або погіршує стан пацієнтів. Проте в окремих випадках дійсно складно провести диференціальну діагностику між різними формами ЛГ (наприклад, рідкісні хвороби легень із незначними порушеннями функції легень та високою ЛГ за даними ехокардіографії (ЕхоКГ); серцева недостатність зі збереженою фракцією викиду, майже нормальними розмірами порожнин серця та високою ЛГ за даними ЕхоКГ) при звичайному обстеженні, і для цього в більшості країн світу існують спеціалізовані центри з діагностики та лікування ЛГ, які мають досвід та необхідне обладнання для проведення додаткових обстежень. В Україні такий центр було створено на базі ДУ «ННЦ «Інститут кардіології імені академіка М.Д. Стражеска» НАМН України» у 2014 році.
Дана стаття є однією із циклу статей, які базуються на клінічних прикладах та мають на меті ознайомити широке коло зацікавлених лікарів з основними принципами діагностики та лікування певних форм ЛГ, які або не потребують призначення специфічної терапії, або ж потребують, але тільки за наявності певних індивідуальних характеристик пацієнта.
Клінічний випадок
Пацієнтка П., 58 років, надійшла до центру легеневої гіпертензії ДУ «ННЦ «Інститут кардіології ім. академіка М.Д. Стражеска» НАМНУ» зі скаргами на виражену задишку при незначному фізичному навантаженні, що значно обмежувала повсякденну фізичну активність (відповідає ІІІ ф.к. ВООЗ), сухий кашель, виражену слабкість і втомлюваність, набряки нижніх кінцівок, серцебиття та перебої в роботі серця при незначному навантаженні, порушення сну через задишку, відчуття «замерзання» фаланг пальців верхніх і нижніх кінцівок, яке турбувало з молодого віку (синдром Рейно). Задишка почала турбувати 10 років тому, коли почалися вікові порушення менструального циклу. Вона поступово прогресувала і більш вираженою стала в грудні 2018 року. При обстеженні за місцем проживання у квітні 2019 року був установлений діагноз ішемічної хвороби серця, гіпертонічної хвороби. Але призначене лікування виявилося неефективним, і пацієнтка була направлена в обласний кардіологічний диспансер, де після ЕхоКГ лікарі запідозрили ідіопатичну легеневу артеріальну гіпертензію (ЛАГ) і направили пацієнтку в центр легеневої гіпертензії ДУ «ННЦ «Інститут кардіології імені акад. М.Д. Стражеска» НАМН України», куди вона поступає в липні 2019 року. Серед супутніх станів: вузловий зоб (за даними ультразвукового обстеження, еутиреоз за даними гормонального обстеження), хірургічне видалення каменів лівої нирки (2016), хірургічне лікування катаракти лівого ока (2017), васкуліт (2014), часті загострення хронічного бронхіту. У квітні 2019 році консультована пульмонологом за місцем проживання після проведення спіральної комп’ютерної томографії: виявили неспецифічну інтерстиціальну пневмонію, дихальну недостатність 2-го ст. Було призначено метилпреднізолон, після якого відчувала незначне полегшення стану.
/40.jpg)
Об’єктивна оцінка стану пацієнтки: стан середньої тяжкості, ожиріння І ст., шкірні покриви ціанотичні (ціаноз збільшувався при фізичному навантаженні), ознаки синдрому Рейно, телеангіоектазії, м’які набряки пальців рук, гомілок, стоп, SpO2 85 %, у легенях крепітуючі хрипи з обох сторін у нижніх відділах, частота дихання — 20, частота серцевих скорочень — 100 уд/хв, систолічний (САТ)/діастолічний (ДАТ) артеріальний тиск — 140/90 мм рт.ст., систолічний шум у т. Боткіна, печінка збільшена (+6 см). Дані загальних лабораторних обстежень наведено в табл. 1. Як видно з табл. 1, у пацієнтки вперше виявлене підвищення рівня глюкози в крові та сечі, і, враховуючи підвищений рівень глікованого гемоглобіну (табл. 2), вперше установлений діагноз цукрового діабету.
/41.jpg)
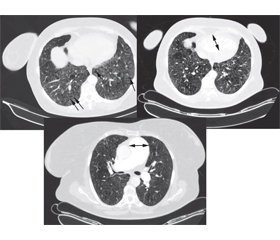
Пацієнтка була обстежена згідно із затвердженим локальним протоколом, що базується на рекомендаціях Європейського товариства кардіологів та Європейського респіраторного товариства (2015) [1], та уніфікованим протоколом, затвердженим МОЗ України (2016) [3]. Шестихвилинний тест продемонстрував значне зниження толерантності до фізичного навантаження: 267 м, задишка за Borg 6 балів, зниження SpО2 до 75 % наприкінці тесту. За даними додаткових лабораторних обстежень (табл. 2), виявлено помірно підвищений рівень N-термінального мозкового натрійуретичного пептиду (NT-proBNP), що говорить про перевантаження правого передсердя. При спіральній комп’ютерній томографії: лімфаденопатія, розширення легеневого ствола до 53 мм, легеневих артерій до 30 мм, легеневі поля помірно емфізематозні, виражена деформація легенево-бронхіальних структур за рахунок пневмофіброзу і пневмосклерозу, ущільнення за типом матового скла з кістозними змінами і сітчастим рисунком над всіма легеневими полями, множинні фіброзно-бульозні зміни, у S5 зліва щільне вогнище до 0,6 см у діаметрі, справа та зліва нечисленні вогнища інтенсивності до 0,3 см у діаметрі, бронхи прохідні, стінки потовщені, деформовані (рис. 1). За даними скринінгової полісомнографії, ознак нічного апное виявлено не було. При звичайній спірометрії: FVC 47 %, FEV1 53 %, FEV1/FVC 113 %.
/41_2.jpg)
Дані ЕхоКГ наведені в табл. 3, з якої видно збільшення розмірів правих відділів серця, які значно переважають над лівими (індекс ексцентричності 1,6 у діастолу та 2,2 у систолу), збільшення діаметра легеневої артерії, значна швидкість регургітації на тристулковому клапані, що свідчить про високий тиск у легеневій артерії (розрахована величина САТ у легеневій артерії — 103 мм рт.ст.). Висока ЛГ була підтверджена даними катетеризації правих відділів серця: середній АТ у легеневій артерії — 77 мм рт.ст., хвилинний об’єм крові (визначено методом термодилюції) — 4,3 л/хв, серцевий індекс — 2,2 л/хв/м2, тиск у правому передсерді — 6 мм рт.ст., тиск заклинювання в легеневій артерії — 11 мм рт.ст., легеневий судинний опір — 1237 дин × с × см–5 (16 одиниць Вуда).
/42.jpg)
Враховуючи дані анамнезу (синдром Рейно з молодого віку, васкуліт, артралгії), об’єктивного обстеження (ознаки Рейно, телеангіоектазії, крепітуючі хрипи з обох сторін) та дані інструментальних методів дослідження (пневмофіброз, наявність прекапілярної ЛГ), у пацієнтки запідозрили захворювання сполучної тканини — можливо, склеродермія. Пацієнтка консультована ревматологом, було додатково рекомендовано провести аналіз крові на скринінг ревматичних хвороб, капіляроскопію, рентгенологічне дослідження кистей. Окрім того, пацієнтка була направлена до пульмонологів для проведення оцінки функції зовнішнього дихання методом бодиплетизмографії та дифузійної здатності легень. Паралельно проводилось лікування: оксигенотерапія (не менше 16 год/добу), діуретична терапія, антидіабетична, продовжена терапія глюкокортикостероїдами згідно зі схемою, відкоригованою ревматологом. Ураховуючи високий рівень тиску в легеневій артерії та те, що можливою причиною ЛГ було захворювання сполучної тканини (група 1 — ЛАГ), при якому призначається специфічна терапія, ex juvantibus було призначено інгаляційний ілопрост (40 мкг/добу) та силденафіл (60 мг/добу). На фоні призначеного лікування стан пацієнтки покращився. Через певні сімейні обставини пацієнтка була виписана на цій терапії ще до отримання результатів рекомендованих вище додаткових досліджень.
Через 3 місяці пацієнтка повторно обстежується в центрі. При цьому суб’єктивно вона не відмічала покращання стану. Ілопрост пацієнтка не прий-мала через його значну вартість та відсутність у регіоні проживання за Програмою забезпечення безкоштовними ліками. Дані 6-хвилинного тесту (246 м, задишка за Borg 10 балів, зниження сатурації О2 до 69 % наприкінці тесту), ЕхоКГ (табл. 3) та рівень NT-proBNP (табл. 2) об’єктивно підтверджували відсутність суттєвого позитивного ефекту від призначеної терапії. Результати проведених додаткових досліджень (скринінговий аналіз крові на ревматичні хвороби, капіляроскопія, рентгенографія кистей) свідчили про малоймовірний попередній діагноз склеродермії. Показники функції зовнішнього дихання (TLC 82,1 %, FEV 50,1 %, FVC 57 %) свідчили про наявність дихальної недостатності ІІ ст. за змішаним типом із переважанням рестриктивного типу. Був проведений консиліум із залученням пульмонологів відділення технологій лікування неспецифічних захворювань легень Національного інституту фтизіатрії та пульмонології ім. Ф.Г. Яновського, які допомогли встановити діагноз рідкісної хвороби легень — лімфангіолейоміоматозу (ЛАМ). Кінцевий діагноз був таким: легенева гіпертензія, асоційована із багатофакторним механізмом виникнення (група 5), ІІІ ст. (за даними катетеризації 2019). Недостатність три-стулкового клапану І ст. Дилатація ствола легеневої артерії. Синусова тахікардія. ІІІ функціональний клас за ВООЗ. Дисліпідемія. Гіперурикемія. Лімфангіолейоміоматоз легень. ЛН ІІ–ІІІ ст. ХЗН І ст. (швидкість клубочкової фільтрації — 99 мл/хв): сечокам’яна хвороба нирок. Стан після видалення конкременту лівої нирки. Цукровий діабет ІІ типу, середнього ступеня тяжкості. Дифузний зоб, еутиреоз. Ожиріння І ст.
Лікування включало призначення оксипрогестерону капронату 12,5% 2 мл внутрішньом’язово один раз на тиждень, препаратів кальцію, оксигенотерапії, підтримуючої дози медролу 6 мг на добу, діуретичної терапії. Щодо лікування легеневої гіпертензії, то прийом інгаляційного ілопросту було відновлено, і на цьому фоні пацієнтка почала почувати себе краще.
Обговорення клінічного випадку
За даними Європейського респіраторного товариства, ЛАМ — це рідке захворювання легень, що зустрічається з частотою 1–5 осіб на 1 млн населення [4, 5]. У США у 2006 році було зареєстровано 230 випадків цього захворювання згідно з реєстром ЛАМ Національного інституту серця, легень і крові [6]. Національний інститут фтизіатрії та пульмонології ім Ф.Г. Яновського повідомляв про 22 випадки даного захворювання [7]. Частіше хворіють жінки дітородного віку. Нашій пацієнтці діагноз ЛАМ установлено у віці 58 років, що не характерно для цього захворювання. Проте клінічна маніфестація хвороби у вигляді задишки була близько 10 років назад, у передменопаузальний період.
Вважається, що ЛАМ у 85 % виникає спорадично, а в 15 % асоціюється з туберозним склерозом (ЛАМ-КТС) [7]. Обидві форми ЛАМ пов’язують із мутаціями генів TSC1 і TSC2, але лише ЛАМ-КТС передається спадково за автосомно-домінантним типом [7]. Етіологія ЛАМ не відома, але важливу роль відіграють естрогенні гормональні порушення. Саме тому хвороба часто загострюється під час вагітності, перед менструаціями, поєднується з лейоміомою матки. Морфологічним субстратом хвороби є проліферація ЛАМ-клітин (атипові гладком’язові й епітеліальні клітини) навколо бронхіол, артерій, вен, лімфатичних судин, у міжальвеолярних перетинках та плеврі. У результаті звужується просвіт дрібних бронхіол із формуванням маленьких кіст, які можуть розриватися, приводячи до рецидивуючих пневмотораксів. Розростання ЛАМ-клітин навколо артерій малого кола кровообігу призводить до зменшення їх просвіту, збільшення легеневого опору, розвитку ЛГ та легеневого серця. Ураження вен призводить до збільшення тиску в капілярах та виникнення геморагій, гемосидерозу та клінічно проявляється кровохарканням. Зміни лімфатичних судин призводять до виникнення хілотораксу та розвитку лімфаденопатії, що спостерігалася в нашої пацієнтки. Нелегеневими проявами ЛАМ є ангіоліпома нирок, менінгіома.
Клінічними проявами ЛАМ є прогресуюча задишка (87 %), пневмоторакс (65 %), кашель (51 %), біль у грудній клітці (34 %), хілоторакс (28 %), кровохаркання (22 %) [8]. За критеріями Європейського респіраторного товариства [5], діагноз ЛАМ може бути визначений, достовірний та можливий. Визначеним ЛАМ вважається: 1) за наявності характерних ознак при спіральній КТ високої здатності та при біопсії легень; 2) наявності характерних ознак при спіральній КТ високої здатності та будь-яких проявів із таких: ангіоміоліпома (нирки), торакальна або перитонеальна хільозна ефузія, лімфангіолейоміома або ураження лімфатичних вузлів ЛАМ-клітинами, наявність туберозного комплексу. Достовірним діагноз вважається: 1) за наявності характерних ознак при спіральній КТ високої здатності та відповідної клінічної історії; 2) наявності сумісних (як ті, що не протирічать) ознак при спіральній КТ високої здатності та будь-чого з такого: ангіоміоліпома (нирки), торакальна або перитонеальна хільозна ефузія. Можливим є діагноз за наявності лише характерних ознак при спіральній КТ високої здатності. Найбільш характерними для ЛАМ КТ-ознаками є наявність тонкостінних кіст різних розмірів (від декількох мм до декількох см) у кількості більше 10. Якщо їх кількість становить 2–10, то ознаки вважаються сумісними. У нашої пацієнтки діагноз був, найімовірніше, достовірним.
Хоча критерії ЛАМ легень розроблені і є специфічними, рідкісність цієї хвороби обумовлює необхідність певного досвіду в постановці цього діагнозу. У нашої пацієнтки ні пульмонологи за місцем проживання, ні спеціалісти з візуалізації не змогли правильно встановити діагноз. Установлення діагнозу в нашому центрі стало можливим лише через співпрацю з Інститутом фтизіатрії та пульмонології ім. Ф.Г. Яновського в рамках єдиного медичного простору Національної академії медичних наук України.
Прогноз при даному захворюванні є не дуже сприятливим. Летальний результат можна очікувати в строки від 3–5 до 15 років. В основному він визначається станом пацієнта: ступенем порушення функції дихання та дифузійною здатністю, за наявності гіпоксемії. Вважається, що емпіричне застосування препаратів прогестерону може сповільнити прогресування хвороби. Окрім того, призначають препарати кальцію через часті прояви остеопорозу. Ефективність глюкокортикостероїдів не доведена. Оксигенотерапія є обов’язковою при сатурації < 90 %. При виражених порушеннях функції легень рекомендована двостороння трансплантація легень.
Поширеність ЛГ при захворюваннях легень достатньо висока. Так, при хронічних обструктивних хворобах ІV стадії середній тиск у легеневій артерії > 20 мм рт.ст. реєструється в 90 % пацієнтів та приблизно 1–5 % хворих мають значну ЛГ (≥ 35 мм рт.ст.) [9]. Поширеність ЛГ, за даними ЕхоКГ, при інтерстиціальних захворюваннях легень (ідіопатичний легеневий фіброз або ідіопатична інтерстиціальна пневмонія) становить 8–15 % на ранній стадії захворювання та > 60 % на пізніх стадіях [10–12]. При цьому майже не існує кореляції між ступенем порушення функції легень та ступенем підвищення тиску в легеневій артерії. При саркоїдозі ЛГ зустрічається в 5,4–74 % пацієнтів, та вона має дуже складний механізм виникнення: фіброзне ремоделювання та облітерація легеневих артерій (як при ідіопатичній ЛАГ), компресія центральних легеневих артерій збільшеними лімфатичними судинами та медіастинальною фіброзною тканиною, венооклюзійне ураження, ураження серця з розвитком дисфункції лівих відділів серця, портальна гіпертензія з розвитком портопульмональної ЛГ [13]. Гемодинамічно при саркоїдозі ЛГ може бути прекапілярною та посткапілярною (при переважанні ураження серця). Саме тому цю ЛГ спеціалісти відносять до групи 5 (із неясним або багатофакторним механізмом розвитку). До цієї ж групи належить ЛГ при ЛАМ легень та легеневому гістіоцитозі. При гістіоцитозі ЛГ частіше є високою, при ЛАМ — м’якою або помірною і ступінь залежить від ступеня ураження паренхіми легень [14, 15].
Діагностика ЛГ включає загальноприйняті обстеження: циркулюючі біомаркери, оцінку функції дихання, ЕхоКГ та спіральну КТ високої здатності. Згідно з даними певних досліджень та положення, затвердженого на 6-му Всесвітньому конгресі з ЛГ 2018 р., рівень мозкового натрійуретичного пептиду має меншу чутливість та специфічність при помірній ЛГ та значною мірою може залежати від стану лівих відділів серця [16–18]. При ідіопатичному легеневому фіброзі та обструктивних захворюваннях легень ЛГ асоціюється з низькою дифузійною здатністю легень, зменшенням толерантності до фізичного навантаження, порушенням газообміну [19, 20]. ЕхоКГ є кращим методом скринінгу на виявлення ЛГ. Розрахований за величиною трикуспідальної регургітації систолічний тиск у легеневій артерії добре корелює з тиском, визначеним при катетеризації правих відділів серця в загальній групі пацієнтів, але індивідуальні коливання можуть бути значними [21]. Тому дуже важливо враховувати й інші показники, що характеризують наявність ЛГ (розміри правих відділів серця, легеневої артерії, співвідношення правих і лівих відділів серця), у тому числі й ті, що визначаються за даними КТ (співвідношення діаметра легеневої артерії та діаметра висхідної аорти (> 1,1) [22–24].
КПС є золотим стандартом для діагностики ЛГ, але вона не є рекомендованою для проведення всім пацієнтам із захворюванням легень, а лише в тих пацієнтів, які мають значне підвищення рівня АТ у легеневій артерії (як це було в нашої пацієнтки), або яким буде проводитися трансплантація легень, або які будуть включатися в спеціальні реєстри/клінічні дослідження, для виключення дисфункції лівих відділів серця та вирішення питання щодо призначення специфічної терапії. Крім того, КПС може обговорюватися: 1) коли клінічне погіршення стану, прогресування обмежень фізичної активності та/або порушення газообміну не відповідають ступеню порушення функції легень; 2) для визначення прогнозу у випадках, коли це є дуже важливим. При захворюваннях легень ЛГ вважається значною, якщо підвищення рівня середнього тиску в легеневій артерії становить > 35 мм рт.ст. або > 25 мм рт.ст. при низькому серцевому індексі (< 2,0 л × хв × м–2) [16].
У більшості клінічних досліджень, які включали пацієнтів із ЛАГ (група 1), критеріями виключення були значні порушення функції легень: TLC < 60–70 %, FEV1 < 55–80 % або FEV1/FVC < 50–70 % [16]. Для цих досліджень рутинно не застосовували візуалізуючі методи для виключення наявності захворювання легень, хоча при паренхіматозних ураженнях показники функції легень часто можуть бути вищими від вказаних критерієв виключення, і, можливо, частина пацієнтів із легеневими захворюваннями також потрапила в ці клінічні дослідження. Окрім того, відомо, що пацієнти з ЛАГ (група 1) часто страждають від хронічних легеневих захворювань (наприклад, при вроджених вадах серця є частими хронічні бронхіти, у тому числі обструктивні), і в цьому разі дві хвороби можуть існувати паралельно. Критерії для диференціації між групами 1 та 3 наведено в табл. 4. У випадках, коли не вдається чітко визначитися з групою ЛГ, рекомендовано направляти пацієнта в референтний центр, де є можливість залучити до обстеження кваліфікованих пульмонологів.
/45.jpg)
Специфічна терапія ЛГ. Дані щодо застосування специфічних препаратів при захворюваннях легень обмежуються результатами нерандомізованих та декількох малочисленних рандомізованих клінічних досліджень, які проводилися із залученням пацієнтів, хворих на інтерстиціальні легеневі хвороби, хронічні обструктивні захворювання легень (ХОЗЛ) та саркоїдоз. У пацієнтів із ХОЗЛ, незважаючи на позитивні зміни гемодинамічних показників при тривалому застосуванні силденафілу та бозентану [25–28], не спостерігалося значного покращання толерантності до фізичного навантаження, клінічної симптоматики та якості життя [25, 26]. Виняток становило дослідження Р. Vitulo зі співавторами, в якому відбулося покращання якості життя на силденафілі [27]. За даними мета-аналізу K. Prins зі співавторами (рис. 2), специфічна терапія достовірно не збільшувала дистанцію при 6-хвилинному тесті [26].
Деякі автори вважають, що вазодилатуюча терапія може погіршувати газообмін через розширення тих судин, які доставляють кров в альвеоли, що не вентилюються, та виникнення «обкрадання» альвеол, що вентилюються [29]. Але це погіршення може бути компенсоване підвищенням серцевого викиду та покращанням доставки кисню в тканини. Окрім того, не в усіх дослідженнях спостерігалося порушення газообміну. Так, у спостереженнях D.J. Lederer зі співавторами та G. Calcaianu зі спів-авторами призначення силденафілу та тадалафілу не погіршувало газообміну [30, 31].
/46.jpg)
Таким чином, хоча загалом специфічна терапія при ХОЗЛ поки що розчаровує (позитивні гемодинамічні зміни не асоціюються з покращанням симптомів та толерантності до фізичного навантаження), певні успішні спостереження свідчать про достовірну користь специфічної терапії при ХОЗЛ та значній ЛГ. При цьому в керівництвах зазначається, що перед тим як рекомендувати специфічне лікування для пацієнтів із ХОЗЛ, необхідно отримати переконливі докази його ефективності в рандомізованих клінічних дослідженнях [16]. У ці дослідження слід включати також пацієнтів із меншою ЛГ (< 35 мм рт.ст.) та низьким серцевим викидом або високим легеневим опором.
Застосування специфічної терапії при ідіопатичній інтерстиціальній пневмонії, а саме амбризентану в дослідженні ARTEMIS та ріоцигуату в дослідженні RISE-IIP, продемонструвало небезпечність цих препаратів для даної категорії хворих. Так, під впливом амбризентану спостерігалося збільшення прогресування захворювання та частоти госпіталізацій із приводу респіраторних порушень [32]. Ріоцигуат показав збільшення смертності та ризику серйозних побічних ефектів [33]. Саме тому амбризентан та ріоцигуат є протипоказаними в пацієнтів з ідіопатичною інтерстиціальною пневмонією. Гемодинамічні покращення спостерігали при застосуванні ріоцигуату та трепростинілу [34, 35], але не на фоні амбризентану та бозентану [10, 36]. У дослідженні STEP-IPF не відмічалося достовірного збільшення дистанції 6-хвилинного тесту на фоні специфічної терапії у хворих з ідіопатичним фіброзом легень [37] на відміну від спостережень (рис. 2) із застосуванням силденафілу, ріоцигуату та трепростинілу [26]. За даними метааналізу, достовірне збільшення дистанції 6-хвилинного тесту спостерігалося лише в нерандомізованих дослідженнях (рис. 2). В рандомізованих дослідженнях зміни були недостовірними [26]. У найбільшому спостереженні, яке включало 151 пацієнта з ідіопатичною інтерстиціальною пневмонією, відповідь на застосування специфічної терапії була такою ж, як і в пацієнтів з ідіопатичною ЛАГ [38.]. Якість життя покращувалася на фоні прийому силденафілу та трепростинілу, але не на інших препаратах [35, 37]. Призначення інгаляційного ілопросту, оксиду азоту та силденафілу не погіршувало газообмін [39–41], тоді як на епопростинілі через «обкрадання» спостерігалося погіршення.
Таким чином, застосування специфічної терапії при інтерстиціальних хворобах легень має суперечливі результати. Наразі ріоцигуат та амбризентан є протипоказаними. Немає даних щодо позитивного впливу інших антагоністів ендотелінових рецепторів. Свідоцтва щодо силденафілу є конфліктними, щодо терапії простаноїдами — частіше позитивними, але обмеженими.
При саркоїдозі [42, 43], гістіоцитозі та ЛАМ легень [44] існує мала кількість даних щодо застосування специфічної терапії (в основному з нерандомізованих досліджень). Єдине рандомізоване дослідження з бозентаном у пацієнтів із саркоїдозом показало покращання гемодинамічних показників, але не толерантності до фізичного навантаження [45]. Наразі рутинне застосування специфічної терапії при цих хворобах не є рекомендованим.
Як видно з вказаних вище даних, специфічна терапія при захворюваннях легень якщо і давала позитивний результат, то він обмежувався лише гемодинамічними показниками або клінічною симптоматикою. Не існує добре спланованих досліджень впливу терапії на виживання. В одному дослідженні покращання виживання відмічалося в пацієнтів із значною ЛГ, але не з м’якою/помірною [46]. На сьогодні існують такі рекомендації щодо застосування специфічної терапії (рис. 3). Якщо в пацієнта мінімальні зміни паренхіми та незначні порушення функції легень, то такий пацієнт може розглядатися як такий, що належить до групи 1 або 5 (багатофакторний або неясний механізм). У першому варіанті специфічна терапія призначається, у другому — пацієнт має бути направлений в експертний центр із ЛГ або пульмонологічний для встановлення діагнозу і ЛГ, і самої хвороби легень. Якщо пацієнт має виражені порушення функції легень та/або виражені зміни паренхіми легень, то він має бути направлений в експертний центр. За наявності м’якої/помірної ЛГ (середній тиск у легеневій артерії — < 35 мм рт.ст.) специфічна терапія не призначається. При тяжкій ЛГ вона може розглядатися в індивідуальному порядку з урахуванням даних клінічних досліджень та етіології захворювання. Так, у нашої пацієнтки відмічалися виражені зміни паренхіми та функції легень та виражена ЛГ.
/47.jpg)
Діагноз ЛАМ легень дозволяє віднести цю ЛГ до 5-ї групи. Одним із патогенетичних механізмів виникнення ЛГ при ЛАМ є проліферація ЛАМ-клітин навколо артерій малого кола кровообігу, що призводить до зменшення їх просвіту, збільшення легеневого опору. Це дуже схоже на зміни, які відбуваються в пацієнтів із ЛАГ. Тому теоретично призначення специфічної терапії, яка має антипроліферативні властивості, могла б покращити стан пацієнтів. Найбільше позитивних ефектів описано для терапії простагландинами, особливо трепростинілу. Але в Україні трепростиніл не зареєстрований. Застосування єдиного доступного в Україні простагландину — інгаляційного ілопросту, який має антитромботичну, протизапальну, антипроліферативну та фібринолітичну дію, супроводжувалося покращанням клінічного стану нашої пацієнтки. Тому продовження даної специфічної терапії ми вважаємо за доцільне. Але проблема полягає в тому, що за положенням МОЗ України пацієнти з ЛГ всіх інших груп, окрім 1-ї та 4-ї, не забезпечуються безкоштовними специфічними препаратами, навіть із такими рідкісними захворюваннями, як ЛАМ легень.
Особливості даного випадку:
— рідкісне захворювання легень, що можливо діагностувати лише за наявності досвіду;
— нетиповим для даного випадку є те, що захворювання діагностовано у відносно пізньому віці пацієнтки (як правило, більшість жінок із таким діагнозом дітородного віку);
— при помірних змінах функції легень спостерігалася значна ЛГ, яка потребувала дообстеження в спеціалізованому центрі;
— незважаючи на діагностоване ураження легень та загальні рекомендації не призначати специфічну терапію при даній формі ЛГ, як виняток були застосовані простагландини, що покращувало клінічний стан пацієнтки.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці даної статті.
Список литературы
1. Galiè N., Humbert M., Vachiery J.L. et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016. 37(1). Р. 67-119 doi: 10.1093/eurheartj/ehv317.
2. Galiè N., McLaughlin V.V., Rubin L.J., Simonneau G. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur. Respir. J. 2019. 53(1). Р. 1802148. Published 2019 Jan 24. doi:10.1183/13993003.02148-2018.
3. Уніфікований клінічний протокол екстреної, первинної, вторинної (спеціалізованої) та третинної (високоспеціалізованої) медичної допомоги. Легенева гіпертензія у дорослих. 2016. Артеріальна гіперетензія. 2016. № 3. С. 51-83.
4. Zhang X., Travis W. Pulmonary Lymphangioleiomyomatosis. Arch. Pathol. Lab. Med. 2010. 134. Р. 1823-1828. doi: 10.1043/2009-0576-RS.1.
5. Johnson S.R., Cordier J.F., Lazor R. et al. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur. Respir. J. 2010. 35. Р. 14-26. doi: 10.1183/09031936.00076209.
6. Ryu J.H., Moss J., Beck G. The NHLBI lymphangioleiomyomatosis registry: Characteristies of 230 patients at enrollment. Am. J. Respir. Crit. Care Med. 2006. 173(1). Р. 105-111. doi: 10.1164/rccm.200409-1298OC.
7. Редкие интерстициальные заболевания легких. Под ред. В.К. Гаврисюка. Киев: ООО «Велес», 2012. 148 с.
8. Harari S., Torre O., Moss J. Lymphangioleiomyomatosis: What do we know and what we looking for? Eur. Respir. Rev. 2011. 20(119). Р. 34-44. doi: 10.1183/09059180.00011010.
9. Chaouat A., Bugnet A.-S., Kadaoui N. et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005. 172. Р. 189-194. doi: 10.1164/rccm.200401-006OC.
10. Raghu G., Nathan S.D., Behr J., Brown K.K. et al. Pulmonary hypertension in idiopathic pulmonary fibrosis with mild to moderate restriction. Eur. Respir. J. 2015. 46. Р. 1370-1377. doi: 10.1183/13993003.01537-2014.
11. Shorr A.F., Wainright J.L., Cors C.S., Lettieri C.J., Nathan S.D. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur. Respir. J. 2007. 30. Р. 715-721. doi: 10.1183/09031936.00107206.
12. Nadrous H.F., Pellikka P.A., Krowka M.J., Swanson K.L. et al. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest. 2005. 128. Р. 2393-2399. doi: 10.1378/chest.128.4.2393.
13. Nunes H., Humbert M., Capron F. et al. Pulmonary hypertension associated with sarcoidosis: mechanisms, haemodynamics and prognosis. Thorax. 2006. 61. Р. 68-74. doi: 10.1136/thx.2005.042838.
14. Fartoukh M., Humbert M., Capron F. et al. Severe pulmonary hypertension in histiocytosis X. Am. J. Respir. Crit. Care Med. 2000. 161. Р. 216-223. doi: 10.1164/ajrccm.161.1.9807024.
15. Le Pavec J., Lorillon G., Jais X. et al. Pulmonary Lan-gerhans cell histiocytosis-associated pulmonary hypertension: clinical characteristics and impact of pulmonary arterial hypertension therapies. Chest. 2012. 142. Р. 1150-1157. doi: 10.1378/chest.11-2490.
16. Nathan S., Barbera J., Gaine S., Harari S. et al. Number 10 in the series «Proceedings of the 6th World Symposium on Pulmonary Hypertension». Ed. by N. Galiè, V.V. McLaughlin, L.J. Rubin and G. Simonneau. Pulmonary hypertension in chro-nic lung disease and hypoxia. Eur. Respir. J. 2019. 53. Р. 1801-1914. https://doi.org/10.1183/13993003.01914-2018.
17. Andersen K.H., Iversen M., Kjaergaard J., Mortensen J., Nielsen-Kudsk J.E. et al. Prevalence, predictors, and survival in pulmonary hypertension related to end-stage chronic obstructive pulmonary disease. J. Heart Lung. Transplant. 2012. 31. Р. 373-380. doi: 10.1016/j.healun.2011.11.020.
18. Leuchte H.H., Baumgartner R.A., Nounou M.E., Vogeser M. et al. Brain natriuretic peptide is a prognostic parameter in chronic lung disease. Am. J. Respir. Crit. Care Med. 2006. 173. Р. 744-750. doi: 10.1164/rccm.200510-1545OC.
19. Furukawa T., Kondoh Y., Taniguchi H., Yagi M. et al. A scoring system to predict the elevation of mean pulmonary arterial pressure in idiopathic pulmonary fibrosis. Eur. Respir. J. 2018. 51. 1701311. doi: 10.1183/13993003.01311-2017.
20. Glaser S., Obst A., Koch B., Henkel B., Grieger A., Felix S.B. et al. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis — the predictive value of exercise capacity and gas exchange efficiency. PLoS One. 2013. 8. e65643. doi: 10.1371/journal.pone.0065643.
21. Greiner S., Jud A., Aurich M., Hess A., Hilbel T. et al. Reliability of noninvasive assessment of systolic pulmonary artery pressure by Doppler echocardiography compared to right heart catheterization: analysis in a large patient population. J. Am. Heart Assoc. 2014. 3. e001103. doi: 10.1161/JAHA.114.001103.
22. D’Andrea A., Stanziola A., Di Palma E. et al. Right ventricular structure and function in idiopathic pulmonary fibrosis with or without pulmonary hypertension. Echocardiography. 2016. 33. P. 57-65. doi: 10.1111/echo.12992.
23. Nowak J., Hudzik B., Jastrze Bski D. et al. Pulmonary hypertension in advanced lung diseases: echocardiography as an important part of patient evaluation for lung transplantation. Clin. Respir. J. 2018. 12. Р. 930-938. doi: 10.1111/crj.12608.
24. Alkukhun L., Wang X.F., Ahmed M.K. et al. Non-invasive screening for pulmonary hypertension in idiopathic pulmonary fibrosis. Respir. Med. 2016. 117. P. 65-72. doi: 10.1016/j.rmed.2016.06.001.
25. Chen X., Tang S., Liu K., Li Q. et al. Therapy in stable chronic obstructive pulmonary disease patients with pulmonary hypertension: a systematic review and meta-analysis. J. Thorac. Dis. 2015. 7. P. 309-319. doi: 10.3978/j.issn.2072-1439.2015.02.08.
26. Prins K.W., Duval S., Markowitz J., Pritzker M., Thenappan T. Chronic use of PAH-specific therapy in World Health Organization Group III Pulmonary Hypertension: a systematic review and meta-analysis. Pulm. Circ. 2017. 7. P. 145-155. doi: 10.1086/690017.
27. Vitulo P., Stanziola A., Confalonieri M. Sildenafil in severe pulmonary hypertension associated with chronic obstructive pulmonary disease: a randomized controlled multicenter clinical trial. J. Heart Lung. Transplant. 2017. 36. P. 166-174. doi: 10.1016/j.healun.2016.04.010.
28. Valerio G., Bracciale P., Grazia D.A. Effect of bosentan upon pulmonary hypertension in chronic obstructive pulmonary disease. Ther. Adv. Respir. Dis. 2009. 3. P. 15-21. doi: 10.1177/1753465808103499.
29. Barbera J.A., Blanco I. Pulmonary hypertension in patients with chronic obstructive pulmonary disease: advances in pathophysiology and management. Drugs. 2009. 69. P. 1153-1171. doi: 10.2165/00003495-200969090-00002.
30. Lederer D.J., Bartels M.N., Schluger N.W. et al. Sildenafil for chronic obstructive pulmonary disease: a randomized crossover trial. COPD. 2012. 9. P. 268-275. doi: 10.3109/15412555.2011.651180.
31. Calcaianu G., Canuet M., Schuller A., Enache I. et al. Pulmonary arterial hypertension-specific drug therapy in COPD patients with severe pulmonary hypertension and mild-to-mode-rate airflow limitation. Respiration. 2016. 91. P. 9-17. doi: 10.1159/000441304.
32. Raghu G., Behr J., Brown K.K. et al. ARTEMIS-IPF Investigators. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann. Intern. Med. 2013. 158. P. 641-649. doi: 10.7326/0003-4819-158-9-201305070-00003.
33. Nathan S.D., Behr J., Collard H.R. Riociguat for idiopathic interstitial pneumonia-associated pulmonary hypertension (RISE-IIP): a randomised, placebo-controlled phase 2b study. Lancet Respir. Med. 2019 Sep. 7(9). P. 780-790. doi: 10.1016/S2213-2600(19)30250-4.
34. Hoeper M.M., Halank M., Wilkens H. et al. Riociguat for interstitial lung disease and pulmonary hypertension: a pilot trial. Eur. Respir. J. 2013. 41. P. 853-860. doi: 10.1183/09031936.00213911.
35. Saggar R., Khanna D., Vaidya A. et al. Changes in right heart haemodynamics and echocardiographic function in an advanced phenotype of pulmonary hypertension and right heart dysfunction associated with pulmonary fibrosis. Thorax. 2014. 69. P. 123-129. doi: 10.1136/thoraxjnl-2013-204150.
36. Corte T.J., Keir G.J., Dimopoulos K. et al. BPHIT Study Group. Bosentan in pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2014. 190. P. 208-217. doi: 10.1164/rccm.201403-0446OC.
37. Zisman D.A., Schwarz M., Anstrom K.J. et al. The Idiopathic Pulmonary Fibrosis Clinical Research Network, A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N. Engl. J. Med. 2010. 363. P. 620-628. doi: 10.1056/NEJMoa1002110.
38. Hoeper M.M., Behr J., Held M. et al. Pulmonary hypertension in patients with chronic fibrosing idiopathic interstitial pneumonias. PLoS One. 2015. 10. e0141911. doi: 10.1371/journal.pone.0141911.
39. Olschewski H., Ghofrani H.A., Walmrath D. et al. Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to lung fibrosis. Am. J. Respir. Crit. Care Med. 1999. 160. P. 600-607. doi: 10.1164/ajrccm.160.2.9810008.
40. Blanco I., Ribas J., Xaubet A. et al. Effects of inhaled nitric oxide at rest and during exercise in idiopathic pulmonary fibrosis. J. Appl. Physiol. 2011. 110. P. 638-645. doi: 10.1152/japplphysiol.01104.2010.
41. Ghofrani H.A., Wiedemann R., Rose F. et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet. 2002. 360. P. 895-900. doi: 10.1016/S0140-6736(02)11024-5.
42. Barnett C.F., Bonura E.J., Nathan S.D. et al. Treatment of sarcoidosis-associated pulmonary hypertension. A two-center experience. Chest. 2009. 135. P. 1455-1461. doi: 10.1378/chest.08-1881.
43. Baughman R.P., Judson M.A., Lower E.E. et al. Inhaled iloprost for sarcoidosis associated pulmonary hypertension. Sarcoidosis. Vasc. Diffuse Lung. Dis. 2009. 26. P. 110-120. PMID: 20560291.
44. Cottin V., Harari S., Humbert M. et al. Groupe d'Etudes et de Recherche sur les Maladies "Orphelines" Pulmonaires (GERM"O"P). Pulmonary hypertension in lymphangioleiomyomatosis: characteristics in 20 patients. Eur. Respir. J. 2012. 40. P. 630-640. doi: 10.1183/09031936.00093111.
45. Baughman R.P., Culver D.A., Cordova F.C. et al. Bosentan for sarcoidosis-associated pulmonary hypertension: a double-blind placebo controlled randomized trial. Chest. 2014. 145. P. 810-817. doi: 10.1378/chest.13-1766.
46. Lange T.J., Baron M., Seiler I., Arzt M., Pfeifer M. Outcome of patients with severe PH due to lung disease with and without targeted therapy. Cardiovasc. Ther. 2014. 32. P. 202-208. doi: 10.1111/1755-5922.12084.

/40.jpg)
/41.jpg)
/41_2.jpg)
/42.jpg)
/45.jpg)
/46.jpg)
/47.jpg)