
Журнал «Здоровье ребенка» Том 15, №4, 2020
Вернуться к номеру
Фенотипи ожиріння у дітей, клінічні прояви й генетичні асоціації
Авторы: Абатуров О.Є., Нікуліна А.O.
ДЗ «Дніпропетровська медична академія Міністерства охорони здоров’я України», м. Дніпро, Україна
Рубрики: Педиатрия/Неонатология
Разделы: Справочник специалиста
Версия для печати
У літературному огляді наведені сучасні уявлення щодо молекулярно-генетичних особливостей, клінічних проявів основних фенотипів ожиріння у дітей. Розвиток ожиріння є результатом дисбалансу між надходженням і розходуванням енергії протягом тривалого періоду. На даний час серед випадків полігенного ожиріння розрізняють два фенотипи, один з яких, що характеризується відсутністю метаболічних порушень, отримав назву «метаболічно здорове ожиріння» (metabolically healthy obese — MHO), а другий, за рахунок наявності метаболічних ускладнень ожиріння, — «метаболічно нездорове ожиріння» (metabolically unhealthy obese — MUO). Основними геномними представниками, які беруть участь в регуляції споживання енергії, є гени греліну, лептину, рецепторів лептину, ген, асоційований з масою та ожирінням, ген рецептора меланокортину 4, глюкагоноподібного пептиду 1, холецистокініну. На відміну від фенотипу MHO, яке переважно зумовлено зміною активності генів, що експресуються в головному мозку, фенотип MUO асоційований з генами, більшість з яких експресуються в периферичних тканинах. Генетичні особливості експресії периферичних тканин, які беруть участь в адипогенезі, зумовлюють розподіл надлишкової жирової тканини: переважне збільшення маси підшкірної жирової тканини призводить до розвитку фенотипу MHO, а надлишок маси вісцеральної та ектопічної жирової тканини — до виникнення фенотипу MUO. Надлишкова маса підшкірного жиру не призводить до системних метаболічних порушень, але являє собою перехідне явище при MHO, у той час як вісцеральне ожиріння й накопичення ектопічного жиру в печінці, підшлунковій залозі, тканинах серця і скелетних м’язах причинно пов’язано з низькорівневим запаленням, інсулінорезистентністю, порушенням обміну глюкози та розвитком серцево-судинних захворювань і притаманно для фенотипу MUO. Відсутність загальноприйнятих критеріїв, призначених для верифікації фенотипу ожиріння, вимагає пошуку нових маркерів ідентифікації порушень різних метаболічних шляхів, які дозволили б вірогідно розрізняти MHO і MUO.
В литературном обзоре приведены современные представления о молекулярно-генетических особенностях, клинических проявлениях основных фенотипов ожирения у детей. Развитие ожирения является результатом дисбаланса между поступлением и расходом энергии в течение длительного периода. В настоящее время среди случаев полигенного ожирения различают два фенотипа, один из которых, характеризующийся отсутствием метаболических нарушений, получил название «метаболически здоровое ожирение» (metabolically healthy obese — MHO), а второй, за счет наличия метаболических осложнений ожирения, — «метаболически нездоровое ожирение» (metabolically unhealthy obese — MUO). Основными геномными представителями, которые участвуют в регуляции потребления энергии, являются гены грелина, лептина, рецепторов лептина, ген, ассоциированный с массой и ожирением, ген рецептора меланокортина 4, глюкагоноподобного пептида 1, холецистокинина. В отличие от фенотипа MHO, которое преимущественно обусловлено изменением активности генов, экспрессируемых в головном мозге, фенотип MUO ассоциирован с генами, большинство из которых экспрессируются в периферических тканях. Генетические особенности экспрессии периферических тканей, участвующих в адипогенезе, обусловливают распределение избыточной жировой ткани: преимущественное увеличение массы подкожной жировой ткани приводит к развитию фенотипа MHO, а избыток массы висцеральной и эктопической жировой ткани — к возникновению фенотипа MUO. Избыточная масса подкожного жира не приводит к системным метаболическим нарушениям, но представляет собой переходное явление при MHO, в то время как висцеральное ожирение и накопление эктопического жира в печени, поджелудочной железе, тканях сердца и скелетных мышцах причинно связано с низкоуровневым воспалением, инсулинорезистентностью, нарушением обмена глюкозы и развитием сердечно-сосудистых заболеваний и характерно для фенотипа MUO. Отсутствие общепринятых критериев, предназначенных для верификации фенотипа ожирения, требует поиска новых маркеров идентификации нарушений различных метаболических путей, которые позволили бы достоверно различать MHO и MUO.
The literature review presents modern ideas about molecular genetic features, clinical manifestations of phenotypes of obesity in children. The development of obesity results from the imbalance between energy intake and expenditure over a long period. Currently, among phenogenic obesity cases, two phenotypes are distinguished: one of which is characterized by the absence of metabolic disorders, called metabolically healthy obese (MHO), and the second, due to the presence of metabolic complications of obesity, is metabolically unhealthy obesity (metabolically unhealthy obese — MUO). The main genomic representatives that participate in the regulation of energy consumption are the genes ghrelin, leptin, leptin receptors, the gene associated with mass and obesity, the melanocortin 4 receptor gene, the glucagon-like peptide 1, and cholecystokinin. In contrast to the MHO phenotype, which is mainly due to changes in the activity of genes expressed in the brain; the MUO phenotype is associated with genes, most of which are mainly expressed in peripheral tissues. Genetic features of the expression of peripheral tissues involved in adipogenesis determine the distribution of excess adipose tissue: a predominant increase in the mass of subcutaneous adipose tissue leads to the development of the MHO phenotype, and excess weight of visceral and ectopic adipose tissue leads to the appearance of the MUO phenotype. Excess weight of subcutaneous fat does not lead to systemic metabolic disorders, but it is a transitional phenomenon in MHO, while visceral obesity and the accumulation of ectopic fat in the liver, pancreas, heart tissues and skeletal muscles are causally associated with low-grade inflammation, insulin resistance, impaired glucose metabolism and the development of cardiovascular disease and is typical for the MUO phenotype. The absence of generally accepted criteria for verifying the phenotype of obesity requires the search for new markers for identifying disorders of various metabolic pathways that would allow us to reliably distinguish MHO and MUO.
ожиріння; фенотипи; генетичні асоціації; діти; огляд
ожирение; фенотипы; генетические ассоциации; дети; обзор
obesity; phenotypes; genetic associations; children; review
Скорочення
Вступ
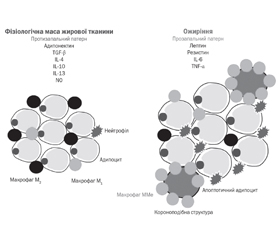
1. Критерії діагностики MHO і MUO
/51.jpg)
2. Розвиток ожиріння й коротка характеристика MHO і MUO
3. Гени, що беруть участь у регуляції прийому їжі
/55.jpg)
/53.jpg)
/54.jpg)
4. Гени периферичних тканин, що асоційовані з порушеннями адипогенезу й ліпідного обміну
Висновки
/56.jpg)
- Абатуров А.Е. Метаболический синдром у детей (лекция). Таврический медико-биологический вестник. 2007. 10. 57-65.
- Абатуров А.Е. Особенности метаболического синдрома у детей. Дитячий лікар. 2011. 4(11). 54-61.
- Бочарова О.В., Теплякова Е.Д. Ожирение у детей и подростков — проблема здравоохранения XXI века. Казанский медицинский журнал. 2020. 101(3). 381-388.
- Васюкова О.В. Ожирение у детей и подростков: критерии диагноза. Ожирение и метаболизм. 2019. 16(1). 70-73.
- Евдокимова Е.Ю., Попова У.Ю. Ожирение у детей. Маркеры метаболического синдрома у детей. Вестник Совета молодых ученых и специалистов Челябинской области. 2017. 1. 2(17). 16-19.
- Alalwan T.A. Phenotypes of Sarcopenic Obesity: Exploring the Effects on Peri-Muscular Fat, the Obesity Paradox, Hormone-Related Responses and the Clinical Implications. Geriatrics (Basel). 2020. 5(1). 8. doi: 10.3390/geriatrics5010008.
- Apalasamy Y.D., Ming M.F., Rampal S., Bulgiba A., Mohamed Z. Association of melanocortin-4 receptor gene polymorphisms with obesity-related parameters in Malaysian Malays. Ann. Hum. Biol. 2013. 40(1). 102-106. doi: 10.3109/03014460.2012.720709.
- Bains V., Kaur H., Badaruddoza B. Association analysis of polymorphisms in LEP (rs7799039 and rs2167270) and LEPR (rs1137101) gene towards the development of type 2 diabetes in North Indian Punjabi population. Gene. 2020. 754. 144846. doi: 10.1016/j.gene.2020.144846.
- Bakhashab S., Filimban N., Altall R.M. et al. The Effect Sizes of PPARγ rs1801282, FTO rs9939609, and MC4R rs2229616 Variants on Type 2 Diabetes Mellitus Risk among the Western Saudi Population: A Cross-Sectional Prospective Study. Genes (Basel). 2020. 11(1). 98.
- Bala C., Craciun A.E., Hancu N. Updating the concept of metabolically healthy obesity. Acta Endocrinol. (Buchar). 2016. 12(2). 197-205. doi: 10.4183/aeb.2016.197.
- Baldini G., Phelan K.D. The melanocortin pathway and control of appetite-progress and therapeutic implications. J. Endocrinol. 2019. 241(1). R1-R33. doi: 10.1530/JOE-18-0596.
- Ben Ali S., Sediri Y., Kallel A. et al. The G3057A LEPR polymorphism is associated with obesity in Tunisian women. Nutr. Metab. Cardiovasc. Dis. 2011. 21(8). 591-596. doi: 10.1016/j.numecd.2009.12.011.
- Berrington de Gonzalez A., Hartge P., Cerhan J.R. et al. Body-mass index and mortality among 1.46 million white adults [published correction appears in N. Engl. J. Med. 2011 Sep 1. 365(9). 869; N. Engl. J. Med. 2010. 363(23). 2211-2219. doi: 10.1056/NEJMoa1000367.
- Berthold H.K., Giannakidou E., Krone W., Mantzoros C.S., Gouni-Berthold I. The Leu72Met polymorphism of the ghrelin gene is associated with a decreased risk for type 2 diabetes. Clin. Chim. Acta. 2009. 399(1–2). 112-116. doi: 10.1016/j.cca.2008.09.022.
- Bing C., Ambye L., Fenger M. et al. Large-scale studies of the Leu72Met polymorphism of the ghrelin gene in relation to the metabolic syndrome and associated quantitative traits. Diabet. Med. 2005. 22(9). 1157-1160. doi: 10.1111/j.1464-5491.2005.01575.x.
- Blüher M. Metabolically Healthy Obesity. Endocr. Rev. 2020. 41(3). doi: 10.1210/endrev/bnaa004.
- Blüher M. The distinction of metabolically “healthy” from “unhealthy” obese individuals. Curr. Opin. Lipidol. 2010. 21(1). 38-43. doi: 10.1097/MOL.0b013e3283346ccc.
- Boumaiza I., Omezzine A., Rejeb J. et al. Relationship between leptin G2548A and leptin receptor Q223R gene polymorphisms and obesity and metabolic syndrome risk in Tunisian volunteers. Genet. Test Mol. Biomarkers. 2012. 16(7). 726-733. doi: 10.1089/gtmb.2011.0324.
- Brandão I., Martins M.J., Monteiro R. Metabolically Healthy Obesity-Heterogeneity in Definitions and Unconventional Factors. Metabolites. 2020. 10(2). 48. doi: 10.3390/metabo10020048.
- Breitfeld J., Kehr S., Müller L. et al. Developmentally Driven Changes in Adipogenesis in Different Fat Depots Are Related to Obesity. Front. Endocrinol. (Lausanne). 2020. 11. 138. doi: 10.3389/fendo.2020.00138.
- Candi E., Tesauro M., Cardillo C. et al. Metabolic profiling of visceral adipose tissue from obese subjects with or without metabolic syndrome. Biochem. J. 2018. 475(5). 1019-1035. doi: 10.1042/BCJ20170604.
- Chait A., den Hartigh L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020. 7. 22. doi: 10.3389/fcvm.2020.00022.
- Childhood overweight and obesity. 2015. http://www.who.int/dietphysicalactivity/childhood/en/.
- Chung S., Kim Y.J., Yang S.J., Lee Y., Lee M. Nutrigenomic Functions of PPARs in Obesogenic Environments. PPAR Res. 2016. 2016. 4794576. doi: 10.1155/2016/4794576.
- Cristancho A.G., Lazar M.A. Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell. Biol. 2011. 12(11). 722-734. doi: 10.1038/nrm3198.
- Crovesy L., Rosado E.L. Interaction between genes involved in energy intake regulation and diet in obesity. Nutrition. 2019. 67-68. 110547. doi: 10.1016/j.nut.2019.06.027.
- Da Fonseca A.C.P., Abreu G.M., Zembrzuski V.M. et al. The association of the fat mass and obesity-associated gene (FTO) rs9939609 polymorphism and the severe obesity in a Brazilian population. Diabetes Metab. Syndr. Obes. 2019. 12. 667-684. doi: 10.2147/DMSO.S199542.
- Da Fonseca A.C.P., Mastronardi C., Johar A., Arcos-Burgos M., Paz-Filho G. Genetics of non-syndromic childhood obesity and the use of high-throughput DNA sequencing technologies. J. Diabetes Complications. 2017. 31(10). 1549-1561. doi: 10.1016/j.jdiacomp.2017.04.026.
- Damanhoury S., Newton A.S., Rashid M., Hartling L., Byrne J.L.S., Ball G.D.C. Defining metabolically healthy obesity in children: a scoping review. Obes. Rev. 2018. 19(11). 1476-1491. doi: 10.1111/obr.12721.
- De Onis M., Blössner M., Borghi E. Global prevalence and trends of overweight and obesity among preschool children. Am. J. Clin. Nutr. 2010. 92(5). 1257-1264. doi: 10.3945/ajcn.2010.29786.
- Després J.P. Body fat distribution and risk of cardiovascular disease: an update. Circulation. 2012. 126(10). 1301-1313. doi: 10.1161/CIRCULATIONAHA. 111.067264.
- Doaei S., Mosavi Jarrahi S.A., Sanjari Moghadam A. et al. The effect of rs9930506 FTO gene polymorphism on obesity risk: a meta-analysis. Biomol. Concepts. 2019. 10(1). 237-242. doi: 10.1515/bmc-2019-0025.
- Drucker D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018. 27(4). 740-756. doi: 10.1016/j.cmet.2018.03.001.
- Etemad A., Ramachandran V., Pishva S.R. et al. Analysis of Gln223Agr polymorphism of Leptin Receptor Gene in type II diabetic mellitus subjects among Malaysians. Int. J. Mol. Sci. 2013. 14(9). 19230-19244. doi: 10.3390/ijms140919230.
- Ferrara D., Montecucco F., Dallegri F., Carbone F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J. Cell Physiol. 2019. 234(12). 21630-21641. doi: 10.1002/jcp.28821.
- Gao C., Langefeld C.D., Ziegler J.T. et al. Genome-Wide Study of Subcutaneous and Visceral Adipose Tissue Reveals Novel Sex-Specific Adiposity Loci in Mexican Americans. Obesity (Silver Spring). 2018. 26(1). 202-212. doi: 10.1002/oby.22074.
- Gao L., Wang L., Yang H., Pan H., Gong F., Zhu H. MC4R Single Nucleotide Polymorphisms Were Associated with Metabolically Healthy and Unhealthy Obesity in Chinese Northern Han Populations. Int. J. Endocrinol. 2019. 2019. 4328909. doi: 10.1155/2019/4328909.
- Garfield A.S., Li C., Madara J.C. et al. A neural basis for melanocortin-4 receptor-regulated appetite. Nat. Neurosci. 2015. 18(6). 863-871. doi: 10.1038/nn.4011.
- Gesta S., Bezy O., Mori M.A., Macotela Y., Lee K.Y., Kahn C.R. Mesodermal developmental gene Tbx15 impairs adipocyte differentiation and mitochondrial respiration. Proc. Natl. Acad. Sci. USA. 2011. 108(7). 2771-2776. doi: 10.1073/pnas.1019704108.
- Gesta S., Blüher M., Yamamoto Y. et al. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc. Natl. Acad. Sci. USA. 2006. 103(17). 6676-6681. doi: 10.1073/pnas.0601752103.
- Goossens G.H., Blaak E.E. Adipose tissue dysfunction and impaired metabolic health in human obesity: a matter of oxygen? Front. Endocrinol. (Lausanne). 2015. 6. 55. doi: 10.3389/fendo.2015.00055.
- Gortan Cappellari G., Barazzoni R. Ghrelin forms in the modulation of energy balance and metabolism. Eat. Weight Disord. 2019. 24(6). 997-1013. doi: 10.1007/s40519-018-0599-6.
- Hamer O.W., Forstner D., Ottinger I. et al. The Pro115Gln polymorphism within the PPAR gamma2 gene has no epidemiological impact on morbid obesity. Exp. Clin. Endocrinol. Diabetes. 2002. 110(5). 230-234. doi: 10.1055/s-2002-33072.
- Heid I.M., Vollmert C., Hinney A. et al. Association of the 103I MC4R allele with decreased body mass in 7937 participants of two population based surveys. J. Med. Genet. 2005. 42(4). Е21. doi: 10.1136/jmg.2004.027011.
- Hill J.H., Solt C., Foster M.T. Obesity associated disease risk: the role of inherent differences and location of adipose depots. Horm. Mol. Biol. Clin. Investig. 2018. 33(2). doi: 10.1515/hmbci-2018-0012.
- Hosseini-Esfahani F., Koochakpoor G., Daneshpour M.S. et al. Mediterranean Dietary Pattern Adherence Modify the Association between FTO Genetic Variations and Obesity Phenotypes. Nutrients. 2017. 9(10). 1064. doi: 10.3390/nu9101064.
- Iacobini C., Pugliese G., Blasetti Fantauzzi C. et al. Metabolically healthy versus metabolically unhealthy obesity. Metabolism. 2019. 92. 51-60. doi: 10.1016/j.metabol.2018.11.009.
- Imaizumi T., Ando M., Nakatochi M. et al. Effect of dietary energy and polymorphisms in BRAP and GHRL on obesity and metabolic traits. Obes. Res. Clin. Pract. 2018. 12(2). 39-48. doi: 10.1016/j.orcp.2016.05.004.
- Jiang N., Li Y., Shu T., Wang J. Cytokines and inflammation in adipogenesis: an updated review. Front. Med. 2019. 13(3). 314-329. doi: 10.1007/s11684-018-0625-0.
- Jiang Y., Mei H., Lin Q. et al. Interaction effects of FTO rs9939609 polymorphism and lifestyle factors on obesity indices in early adolescence. Obes. Res. Clin. Pract. 2019. 13(4). 352-357. doi: 10.1016/j.orcp.2019.06.004.
- Joatar F.E., Al Qarni A.A., Ali M.E. et al. Leu72Met and Other Intronic Polymorphisms in the GHRL and GHSR Genes Are Not Associated with Type 2 Diabetes Mellitus, Insulin Resistance, or Serum Ghrelin Levels in a Saudi Population. Endocrinol. Metab. (Seoul). 2017. 32(3). 360-369. doi: 10.3803/EnM.2017.32.3.360.
- Kahn C.R., Wang G., Lee K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Invest. 2019. 129(10). 3990-4000. doi: 10.1172/JCI129187.
- Karastergiou K., Fried S.K., Xie H. et al. Distinct developmental signatures of human abdominal and gluteal subcutaneous adipose tissue depots. J. Clin. Endocrinol. Metab. 2013. 98(1). 362-371. doi: 10.1210/jc.2012-2953.
- Karpe F., Pinnick K.E. Biology of upper-body and lower-body adipose tissue-link to whole-body phenotypes. Nat. Rev. Endocrinol. 2015. 11(2). 90-100. doi: 10.1038/nrendo.2014.185.
- Kilpeläinen T.O., Zillikens M.C., Stančákova A. et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat. Genet. 2011. 43(8). 753-760. doi: 10.1038/ng.866.
- Kodama N., Tahara N., Tahara A. et al. Effects of pioglitazone on visceral fat metabolic activity in impaired glucose tolerance or type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2013. 98(11). 4438-4445. doi: 10.1210/jc.2013-2920.
- Komşu-Ornek Z., Demirel F., Dursun A. et al. Leptin receptor gene Gln223Arg polymorphism is not associated with obesity and metabolic syndrome in Turkish children. Turk. J. Pediatr. 2012. 54(1). 20-24.
- Kratz M., Coats B.R., Hisert K.B. et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014. 20(4). 614-625. doi: 10.1016/j. cmet.2014.08.010.
- Lauria F., Siani A., Picó C. et al. A Common Variant and the Transcript Levels of MC4R Gene Are Associated With Adiposity in Children: The IDEFICS Study. J. Clin. Endocrinol. Metab. 2016. 101(11). 4229-4236. doi: 10.1210/jc.2016-1992.
- Lee K.Y., Yamamoto Y., Boucher J. et al. Shox2 is a molecular determinant of depot-specific adipocyte function [published correction appears in Proc. Natl. Acad. Sci. USA. 2016 Apr 19. 113(16). E2347; Proc. Natl. Acad. Sci. USA. 2013. 110(28). 11409-11414. doi: 10.1073/pnas.1310331110.
- Li J., Niu X., Li J., Wang Q. Association of PPARG Gene Polymorphisms Pro12Ala with Type 2 Diabetes Mellitus: A Meta-analysis. Curr. Diabetes Rev. 2019. 15(4). 277-283. doi: 10.2174/1573399814666180912130401.
- Lin H., Zhang L., Zheng R. et al. The prevalence, metabolic risk and effects of lifestyle intervention for metabolically healthy obesity: a systematic review and meta-analysis: A PRISMA-compliant article. Medicine (Baltimore). 2017. 96(47). Е8838. doi: 10.1097/MD.0000000000008838.
- Liu L., Fan Q., Zhang F. et al. A Genomewide Integrative Analysis of GWAS and eQTLs Data Identifies Multiple Genes and Gene Sets Associated with Obesity. Biomed. Res. Int. 2018. 2018. 3848560. doi: 10.1155/2018/3848560.
- Liu P., Shi H., Huang C. et al. Association of LEP A19G polymorphism with cancer risk: a systematic review and pooled analysis. Tumour Biol. 2014. 35(8). 8133-8141. doi: 10.1007/s13277-014-2088-5.
- Loh N.Y., Minchin J.E.N., Pinnick K.E. et al. RSPO3 impacts body fat distribution and regulates adipose cell biology in vitro. Nat. Commun. 2020. 11(1). 2797. doi: 10.1038/s41467-020-16592-z.
- Lonardo A., Mantovani A., Lugari S. et al. Epidemiology and pathophysiology of the association between NAFLD and metabolically healthy or metabolically unhealthy obesity. Ann. Hepatol. 2020. S1665-2681(20)30023-5. doi: 10.1016/j.aohep.2020.03.001.
- Makris M.C., Alexandrou A., Papatsoutsos E.G. et al. Ghrelin and Obesity: Identifying Gaps and Dispelling Myths. A Reappraisal. In Vivo. 2017. 31(6). 1047-1050. doi: 10.21873/invivo.11168.
- Manolopoulos K.N., Karpe F., Frayn K.N. Gluteofemoral body fat as a determinant of metabolic health. Int. J. Obes. (Lond.). 2010. 34(6). 949-959. doi: 10.1038/ijo.2009.286.
- McLaughlin T., Lamendola C., Coghlan N. et al. Subcutaneous adipose cell size and distribution: relationship to insulin resistance and body fat. Obesity (Silver Spring). 2014. 22(3). 673-680. doi: 10.1002/oby.20209.
- Mota de Sá P., Richard A.J., Hang H., Stephens J.M. Transcriptional Regulation of Adipogenesis. Compr. Physiol. 2017. 7(2). 635-674. doi: 10.1002/cphy.c160022.
- Neeland I.J., Ross R., Després J.P. et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: a position statement. Lancet Diabetes Endocrinol. 2019. 7(9). 715-725. doi: 10.1016/S2213-8587(19)30084-1.
- Parikh H., Groop L. Candidate genes for type 2 diabetes. Rev. Endocr. Metab. Disord. 2004. 5(2). 151-176. doi: 10.1023/B:REMD.0000021437.46773.26.
- Peer N., Balakrishna Y., Durao S. Screening for type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2020. 5(5). CD005266. doi: 10.1002/14651858.CD005266.pub2.
- Perrini S., Ficarella R., Picardi E. et al. Differences in gene expression and cytokine release profiles highlight the heterogeneity of distinct subsets of adipose tissue-derived stem cells in the subcutaneous and visceral adipose tissue in humans. PLoS One. 2013. 8(3). Е57892. doi: 10.1371/journal.pone.0057892.
- Prastowo N.A., Haryono I.R. Elevated blood pressure and its relationship with bodyweight and anthropometric measurements among 8-11-year-old Indonesian school children. J. Public Health Res. 2020. 9(1). 1723. doi: 10.4081/jphr.2020.1723.
- Rana S., Bhatti A.A. Association and interaction of the FTO rs1421085 with overweight/obesity in a sample of Pakistani individuals. Eat Weight Disord. 2019. 10.1007/s40519-019-00765-x. doi: 10.1007/s40519-019-00765-x.
- Reilly S.M., Saltiel A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017. 13(11). 633-643. doi: 10.1038/nrendo.2017.90.
- Schleinitz D., Böttcher Y., Blüher M. et al. The genetics of fat distribution. Diabetologia. 2014. 57(7). 1276-1286. doi: 10.1007/s00125-014-3214-z.
- Speakman J.R. The Fat Mass and Obesity Related’ (FTO) gene: Mechanisms of Impact on Obesity and Energy Balance. Curr. Obes. Rep. 2015. 4(1). 73-91. doi: 10.1007/s13679-015-0143-1.
- Srivastava A., Mittal B., Prakash J. et al. Evaluation of MC4R [rs17782313, rs17700633], AGRP [rs3412352] and POMC [rs1042571] Polymorphisms with Obesity in Northern India. Oman. Med. J. 2014. 29(2). 114-118. doi: 10.5001/omj.2014.28.
- Stefan N., Häring H.U., Cusi K. Non-alcoholic fatty liver disease: causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019. 7(4). 313-324. doi: 10.1016/S2213-8587(18)30154-2.
- Steinert R.E., Feinle-Bisset C., Asarian L. et al. Ghrelin, CCK, GLP-1, and PYY(3-36): Secretory Controls and Physiological Roles in Eating and Glycemia in Health, Obesity, and After RYGB. Physiol. Rev. 2017. 97(1). 411-463. doi: 10.1152/physrev.00031.2014.
- Sukhonthachit P., Aekplakorn W., Hudthagosol C. et al. The association between obesity and blood pressure in Thai public school children. BMC Public Health. 2014. 14. 729. doi: 10.1186/1471-2458-14-729.
- Tang Y., Jin B., Zhou L. et al. MeQTL analysis of childhood obesity links epigenetics with a risk SNP rs17782313 near MC4R from meta-analysis. Oncotarget. 2017. 8(2). 2800-2806. doi: 10.18632/oncotarget.13742.
- Tatusova T., Ciufo S., Fedorov B. et al. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2015. 43(7). 3872. doi: 10.1093/nar/gkv278.
- Tchkonia T., Giorgadze N., Pirtskhalava T. et al. Fat depot-specific characteristics are retained in strains derived from single human preadipocytes. Diabetes. 2006. 55(9). 2571-2578. doi: 10.2337/db06-0540.
- Tchkonia T., Lenburg M., Thomou T. et al. Identification of depot-specific human fat cell progenitors through distinct expression profiles and developmental gene patterns. Am. J. Physiol. Endocrinol. Metab. 2007. 292(1). E298-E307. doi: 10.1152/ajpendo.00202.2006.
- Tsatsoulis A., Paschou S.A. Metabolically Healthy Obesity: Criteria, Epidemiology, Controversies, and Consequences. Curr. Obes. Rep. 2020 Jun. 9(2). 109-120. doi: 10.1007/s13679-020-00375-0.
- Turcotte M., Abadi A., Peralta-Romero J. et al. Genetic contribution to waist-to-hip ratio in Mexican children and adolescents based on 12 loci validated in European adults. Int. J. Obes. (Lond.). 2019. 43(1). 13-22. doi: 10.1038/s41366-018-0055-8.
- Vecchié A., Dallegri F., Carbone F. et al. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur. J. Intern. Med. 2018. 48. 6-17. doi: 10.1016/j.ejim.2017.10.020.
- Vorotnikov A.V., Stafeev I.S., Menshikov M.Y. et al. Latent Inflammation and Defect in Adipocyte Renewal as a Mechanism of Obesity-Associated Insulin Resistance. Biochemistry (Mosc.). 2019. 84(11). 1329-1345. doi: 10.1134/S0006297919110099.
- Vukovic R., Dos Santos T.J., Ybarra M. et al. Children With Metabolically Healthy Obesity: A Review. Front. Endocrinol. (Lausanne). 2019. 10. 865. doi: 10.3389/fendo.2019.00865.
- Wan R., Ding Z., Xia S. et al. Effects of PPARγ2 Pro12Ala variant on adipocyte phenotype dependent of DHA. Diabetes Metab. Syndr. Obes. 2019. 12. 2273-2279. doi: 10.2147/DMSO.S214526.
- Wang Q.A., Tao C., Gupta R.K. et al. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013. 19(10). 1338-1344. doi: 10.1038/nm.3324.
- Xi B., Chandak G.R., Shen Y. et al. Association between common polymorphism near the MC4R gene and obesity risk: a systematic review and meta-analysis. PLoS One. 2012. 7(9). Е45731. doi: 10.1371/journal. pone.0045731.
- Yan J., Wang X., Tao H. et al. Lack of association between leptin G-2548A polymorphisms and obesity risk: Evidence based on a meta-analysis. Obes. Res. Clin. Pract. 2015. 9(4). 389-397. doi: 10.1016/j.orcp.2015.01.002.
- Yang J., Loos R.J., Powell J.E. et al. FTO genotype is associated with phenotypic variability of body mass index. Nature. 2012. 490(7419). 267-272. doi: 10.1038/nature11401.
- Yang L.K., Tao Y.X. Biased signaling at neural melanocortin receptors in regulation of energy homeostasis. Biochim. Biophys. Acta Mol. Basis Dis. 2017. 1863(10 Pt. A). 2486-2495. doi: 10.1016/j.bbadis.2017.04.010.
- Yang X., Smith U. Adipose tissue distribution and risk of metabolic disease: does thiazolidinedione-induced adipose tissue redistribution provide a clue to the answer? Diabetologia. 2007. 50(6). 1127-1139. doi: 10.1007/s00125-007-0640-1.
- You Y., Yu Y., Wu Y. et al. Association Study between Ghrelin Gene Polymorphism and Metabolic Syndrome in a Han Chinese Population. Clin. Lab. 2017. 63(1). 175-181. doi: 10.7754/Clin.Lab.2016.160715.
- Yu K., Li L., Zhang L., Guo L., Wang C. Association between MC4R rs17782313 genotype and obesity: A meta-analysis. Gene. 2020. 733. 144372. doi: 10.1016/j.gene.2020.144372.
- Zatterale F., Longo M., Naderi J. et al. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020. 10. 1607. doi: 10.3389/fphys.2019.01607.
- Zhao X., Yang Y., Sun B.F. et al. FTO and obesity: mechanisms of association. Curr. Diab. Rep. 2014. 14(5). 486. doi: 10.1007/s11892-014-0486-0.