Резюме
Останніми десятиліттями виявлено особливі моногенні форми артеріальної гіпертензії (АГ), спричинені специфічними шляхами, обумовленими порушеними рідкісними мутаціями в окремих генах, що призводить до раннього та тяжкого фенотипу АГ. На сьогодні відомо про не менше 37 генів, порушення функції яких чітко супроводжується порушенням регуляції артеріального тиску (АТ), що покращує наше розуміння як механізмів розвитку, так і лікування АГ. Генетичні причини вторинної гіпертензії, як правило, обумовлені порушенням одного гена. Відкриття генів, відповідальних за моногенні форми АГ, розкрило важливу роль нирок та надниркових залоз у регуляції рівня АТ. Більшість із цих синдромів обумовлені мутаціями, які приводять до посилення або втрати функції, що, зі свого боку, призводить до зміни вмісту мінералокортикоїдів, глюкокортикоїдів або активації симпатичних шляхів. Моногенні форми АГ часто призводять до тяжких форм гіпертензії, електролітних та гормональних порушень, супроводжуються резистентністю до препаратів, а також часто до більшого ризику серцево-судинних подій та передчасної смерті. У лекції дані рекомендації щодо генетичного тестування при АГ, алгоритм діагностичного обстеження пацієнта з підозрою на моногенну форму АГ, а також наведені особливості клінічного перебігу та лікування основних відомих форм моногенної АГ.
За последние десятилетия обнаружены особые моногенные формы артериальной гипертензии (АГ), вызванные специфическими путями, обусловленными нарушенными редкими мутациями в отдельных генах, что приводит к раннему и тяжелому фенотипу АГ. На сегодняшний день известно о не менее 37 генах, нарушение функции которых четко сопровождается нарушением регуляции артериального давления (АД), что улучшает наше понимание как механизмов развития, так и лечения АГ. Генетические причины вторичной гипертензии, как правило, обусловлены нарушением одного гена. Открытие генов, ответственных за моногенные формы АГ, раскрыло важную роль почек и надпочечников в регуляции уровня АД. Большинство из этих синдромов обусловлены мутациями, которые приводят к усилению или потере функции, которые, в свою очередь, приводят к изменению содержания минералокортикоидов, глюкокортикоидов или активации симпатических путей. Моногенные формы АГ часто приводят к тяжелым формам гипертензии, электролитным и гормональным нарушениям, сопровождающимся резистентностью к препаратам, а также часто к большему риску сердечно-сосудистых событий и преждевременной смерти. В лекции даны рекомендации по генетическому тестированию при АГ, алгоритм диагностического обследования пациента с подозрением на моногенную форму АГ, а также приведены особенности клинического течения и лечения основных известных форм моногенных АГ.
In recent decades, some monogenic forms of arterial hypertension have been identified. They were caused by specific pathways due to rare disruptive mutations in individual genes that determined an early and severe hypertension phenotype. At least 37 genes are known, the disruption of which is clearly accompanied by a dysfunction of the blood pressure regulation. This knowledge improves our understanding of both the mechanisms of development and treatment of hypertension. The genetic causes of secondary hypertension are usually due to a single gene disorder. The discovery of genes responsible for monogenic forms of hypertension revealed the role of the kidneys and adrenal glands as important players in the regulation of blood pressure. Most of these syndromes are caused by mutations that lead to increased of the function or its loss, which in turn leads to changes in the content of mineralocorticoids, glucocorticoids or sympathetic pathway activation. Monogenic forms of hypertension often lead to severe forms of hypertension, electrolyte and hormonal disorders, accompanied by drug resistance, and often cause a higher risk of cardiovascular events and premature death. The lecture provides recommendations for genetic testing in hypertension, an algorithm for diagnostic examination of a patient with suspected monogenic form of hypertension, as well as features of the clinical course and treatment of the main known forms of monogenic hypertension.
Багатофакторний характер регуляції артеріального тиску (АТ) ілюструється мозаїчною теорією артеріальної гіпертензії (АГ), запропонованою I. Page в 1960 р., яка стверджувала, що есенціальна гіпертензія — це не одне захворювання, а кілька різних захворювань із різним походженням та розвитком, які включають взаємодію між генетикою, навколишнім середовищем та адаптацією, нервові, механічні та гормональні порушення [1]. У той же час були виявлені особливі моногенні форми АГ, спричинені специфічними шляхами, обумовленими порушеними рідкісними мутаціями в окремих генах, що призводить до раннього та тяжкого фенотипу АГ. На сьогодні відомо про не менше 37 генів, порушення функції яких чітко супроводжується порушенням регуляції АТ, що покращує наше розуміння як механізмів розвитку, так і лікування АГ [2]. Генетичні причини вторинної гіпертензії, як правило, обумовлені порушенням одного гена [3]. Відкриття генів, відповідальних за моногенні форми АГ, розкрило важливу роль нирок та надниркових залоз у регуляції рівня АТ. Більшість із цих синдромів обумовлені мутаціями, які призводять до посилення або втрати функції, які, зі свого боку, призводять до зміни вмісту мінералокортикоїдів, глюкокортикоїдів або активації симпатичних шляхів. Моногенні форми АГ часто призводять до тяжких форм гіпертензії, електролітних та гормональних порушень, супроводжуються резистентністю до препаратів, а також часто до більшого ризику серцево-судинних подій та передчасної смерті.
Моногенна АГ може включати такі механізми: а) посилений транспорт Na+, викликаний впливом мінералокортикоїдів, включаючи синдроми явного надлишку мінералокортикоїдів, глюкокортикоїд-лікований альдостеронізм та вроджену гіперплазію надниркових залоз через дефіцит 11β-гідроксилази або 17α-гідроксилази; б) збільшення транспорту Na+, незалежного від рівня мінералокортикоїдів, включаючи синдроми Ліддла та Гордона [4].
Рекомендації щодо генетичного тестування при АГ були надані Європейським товариством гіпертензії [5]. У табл. 1 подані генетичні причини вторинної гіпертензії, що є рідкісними, але важливими, тому що виявлення причини може визначати специфічну медикаментозну терапію. Загальні особливості цих генетичних розладів полягають у тому, що вони, як правило, обумовлюють наявність гіпертензії в дітей, підлітків або осіб молодого віку, а також більшість моногенних розладів викликають підвищення АТ через збільшення реабсорбції натрію в ниркових канальцях. Вони зазвичай асоціюються зі зниженою концентрацією реніну в плазмі крові або активністю реніну в плазмі, що є нетиповим для молодих пацієнтів, а особливо для тих, хто приймає антигіпертензивні засоби (наприклад, блокатори ренін-ангіотензинової системи (РАС), блокатори кальцієвих каналів (БКК) або діуретики), які, як очікується, збільшують концентрацію реніну в плазмі крові або активність реніну в плазмі. Таким чином, виявлення зниженої концентрації реніну в плазмі крові або активності реніну в плазмі, особливо під час прийому блокаторів РАС, БКК або діуретиків, повинно викликати підозру на вторинну гіпертензію через стан, що супроводжується затримкою натрію та води в організмі людини. Важливо, що, зокрема, бета-блокатори, нестероїдні протизапальні препарати, альфа-метилдопа або клонідин пригнічують концентрацію реніну в плазмі крові або активність реніну в плазмі. Прийом цих препаратів слід припинити (якщо це клінічно можливо!) принаймні за 2 тижні до визначення концентрації реніну в плазмі крові або активності реніну в плазмі. Діагностика ґрунтується на плановому фізичному обстеженні, вимірі артеріального тиску та лабораторному аналізі реніну, альдостерону, кортизолу та калію. Генетичне тестування корисне для підтвердження діагнозу та диференціальної діагностики. При диференціальній діагностиці необхідно враховувати артеріальну гіпертензію на тлі паренхіматозної хвороби нирок, стеноз ниркової артерії, новоутворення надниркових залоз, гіпертиреоз, зловживання алкоголем та надмірне споживання дієтичної солі.
/34.jpg)
У Європейських рекомендаціях зазначено, що генетичне тестування повинно розглядатися в спеціалізованих центрах (рівень доказів В, клас рекомендацій ІІа) для пацієнтів, у яких є підозри на рідкісні моногенні причини вторинної гіпертензії, або для тих, хто має феохромоцитому. Рутинне генетичне тестування при гіпертензії не рекомендоване (рівень доказів С, клас рекомендацій ІІІ) [5].
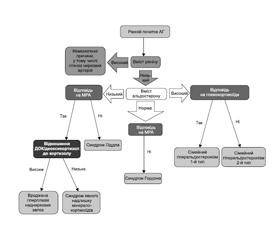
Діагностичний алгоритм щодо виявлення специфічних моногенних форм АГ наведений на рис. 1.
Сімейний гіперальдостеронізм 1-го типу або глюкокортикоїд-супресивна (лікована) гіперплазія є рідкісною формою спадкового захворювання з автосомно-домінантним типом передачі і характеризується станом, коли симптоми альдостеронізму зникають при застосуванні глюкокортикостероїдів у низьких дозах, наприклад дексаметазону. У хворих відзначають підвищену чутливість до альдостеронстимулюючої дії адренокортикотропного гормона. Показано, що причиною захворювання є аномальний ген, що виникає при нерівномірному розподілі хромосоми 8q, у результаті чого порушується порядок кодування альдостеронсинтази.
В організмі це призводить до підвищення продукції альдостерону, а також конверсії кортизолу в гідрокси- й оксикортизол. Захворювання може бути діагностоване за допомогою молекулярно-генетичного аналізу, і при підвищенні доступності технологій у майбутньому тестування слід проводити всім хворим із сімейною формою первинного гіперальдостеронізму.
Для сімейного гіперальдостеронізму 2-го типу причинний ген ще не ідентифікований. Сімейний гіперальдостеронізм 2-го типу дуже складно відрізнити від спорадичного первинного альдостеронізму, за винятком наявності більшої кількості хворих на цю недугу в одній родині. Діагноз цього стану установлюють після виключення інших сімейних форм первинного альдостеронізму [6].
Нещодавно описано сімейний гіперальдостеронізм 3-го типу, спричинений мутаціями гена KCNJ5, що кодує внутрішньовипрямлений калієвий канал Kir3.4. Описані мутації розташовані поблизу або всередині фільтра селективності каналу і відповідають за втрату іонної селективності, що призводить до надходження Na+, деполяризації мембранних клітин, відкриття залежних від напруження кальцієвих каналів, збільшення внутрішньоклітинної концентрації кальцію та вироблення альдостерону. У пацієнтів спостерігається виражена гіпокаліємія, АГ, що складно піддається лікуванню, та двостороння гіперплазія надниркових залоз. Здебільшого необхідна двобічна адреналектомія [4, 6].
Іншою формою спадкового первинного мінералокортицизму є так званий адреногенітальний синдром або вроджена гіперплазія надниркової залози, що становить групу захворювань, для яких характерний дефект ензимів стероїдного синтезу. АГ виникає при дефіциті 11β- або 17α-гідроксилази, за наявності яких відзначають накопичення активних проміжних продуктів мінералокортикоїдного синтезу. Перший тип супроводжується вірилізацією, при цьому часто відзначають вроджені зміни статевих органів: у хлопчиків — макрогенітосомію, у дівчаток — урогенітальний синус або гіпертрофію клітора. Другий тип, навпаки, — затримкою розвитку статевих органів і вторинних статевих ознак (псевдогермафродитизм). АГ 1-го та 2-го ступенів спостерігається приблизно в 60 % випадків на момент установлення діагнозу.
Як і при глюкокортикоїд-лікованій гіперплазії, так і при вродженій гіперплазії надниркових залоз цілями терапії є заміщення недостатньої секреції кортизолу та зменшення надмірної секреції андрогенів та мінералокортикоїдів. Лікування полягає в короткому введенні глюкокортикоїдів у вигляді гідрокортизону в дітей (10–20 мг/м2/добу) у два-три прийоми та глюкокортикоїдів тривалої дії у вигляді преднізолону або дексаметазону (2,5–7,5 мг/добу та 0,25–0,5 мг/добу відповідно) у дорослих. Після початку адекватного лікування глюкокортикоїдами рівень дезоксикортикостерону та реніну зазвичай нормалізується, тоді як для контролю АГ можуть знадобитися додаткові антигіпертензивні препарати [7]. Амілорид є ефективним у боротьбі з гіпокаліємією, проте для контролю АТ не є таким потужним, як спіронолактон. Призначаються й інші стандартні антигіпертензивні засоби в комбінації.
Ще одним рідкісним захворюванням із цієї групи АГ є синдром Ліддла, описаний як спадкове захворювання з автосомно-домінантним типом успадкування в 1963 р. Це захворювання є одною з найбільш поширених так званих моногенних причин АГ. На сьогодні вважають, що захворювання є наслідком генетичного дефекту β- або γ-субодиниці натрієвого каналу епітелію, який призводить до активації каналу, підвищення реабсорбції натрію (підвищення АТ) й екскреції калію (гіпокаліємія). У хворих на фоні раннього розвитку АГ відзначають всі ознаки гіперпродукції мінералокортикоїдів, але рівень альдостерону залишається нормальним. У крові не виявляється підвищення ні однієї з відомих мінералокортикоїдактивних субстанцій. Призначення антагоністів або блокаторів синтезу альдостерону не призводить до зниження АТ. На сьогодні виявлено близько 30 різних гетерозиготних мутацій, що викликають синдром Ліддла, що трапляються як у сімейних, так і в спорадичних випадках: у літературі повідомляється про близько 100 сімейних або спорадичних випадків.
Діагноз синдрому Ліддла базується на клінічній підозрі з урахуванням описаних ознак та симптомів; однак добра реакція на 4-тижневе лікування тріамтереном (100 мг/добу) або амілоридом (10 мг/добу) для корекції гіпокаліємії та гіпертонії є наріжним каменем. Найбільш точним інструментом для ідентифікації синдрому Ліддла є генетичне дослідження з прямим секвенуванням залучених генів та знаходження відомої мутації [4, 8].
Синдром Ліддла асоціюється з підвищеним ризиком серцево-судинних подій (передчасний інсульт, інфаркт міокарда, раптова смерть), пов’язаних не тільки з неправильно діагностованою та недолікованою АГ, але також із дисфункцією ендотелію. Тому синдром Ліддла слід підозрювати у всіх пацієнтів із гіпертонічною хворобою на ранніх стадіях незалежно від вмісту калію, навіть за відсутності сімейного анамнезу, оскільки можуть виникати мутації de novo.
Спостереження показали, що застосування діуретиків, які пригнічують реабсорбцію натрію в дистальних канальцях (амілорид, тріамтерен), може нормалізувати як АТ, так і гіпокаліємію. Так само можна розглядати призначення й інших стандартних антигіпертензивних засобів у комбінації.
Gordon та Hodsman у 1964 р. повідомили про випадок АГ та тяжкої гіперкаліємії зі спадковим характерним фенотипом, метаболічним ацидозом, симптомами періодичного паралічу (через підвищений рівень калію) та зі збереженою функцією нирок. Псевдогіпоальдостеронізм 2-го типу, або синдром Гордона — рідкісна сімейна моногенна форма гіпертонії з автосомно-домінантною ознакою (хоча нещодавно він був описаний із рецесивним успадкуванням). Вона характеризується головним чином характерною ознакою тяжкої гіперкаліємії (досягає 8–9 ммоль/л), що відрізняє синдром Гордона від усіх інших рідкісних синдромних моногенних форм гіпертензії з низьким вмістом реніну. Виділяють три типи синдрому Гордона: тип А — автосомно-домінантний, що впливає на сімейство серин-треонінкіназ WNK (WNK1 та WNK4). Тип B обумовлений мутацією гена WNK4 на хромосомі 17q21.2 із втратою функції, тоді як тип C є результатом мутації гена WNK1 на хромосомі 12p12.3 із посиленням функції [4]. Пацієнти із синдромом Гордона реагують на агресивне дієтичне обмеження солі або невеликі дози тіазидних діуретиків, що передбачає активацію чутливого до тіазидів котранспортера Na/Cl у дистальному нефроні.
Синдром Геллера, що також називають конститутивною активацією мінералокортикоїдного рецептора, є результатом мутації посилення функції хромосоми 4q31. Синдром Геллера має автосомно-домінантний характер успадкування. Ця мутація всередині рецептора мінералокортикоїдів змушує його залишатися конститутивно активним через зміни на ділянках рецепторів, що призводять до зміни специфічності до стероїдних гормонів. Стероїдні гормони, включаючи прогестерон, згодом діють як агоністи мінералокортикоїдного рецептора на відміну від їх нормального антагонізму. АГ починається до 20 років. Важливо розпізнати цю патологію в підлітковому віці, оскільки вагітність може сильно посилити АГ через підвищений вміст прогестерону. Це спричиняє активацію мінералокортикоїдних рецепторів, пригнічується вміст як альдостерону, так і реніну. У таких хворих рівень калію є нормальним. Чіткий діагноз можна встановити за допомогою генетичного тестування на генні мутації в мінералокортикоїдних рецепторах. Лікування не схоже на багато інших причин моногенної гіпертензії, оскільки спіронолактон протипоказаний і погіршить захворювання. Основою лікування під час вагітності є виношування плода. Оптимальне лікування невагітних жінок та чоловіків є недостатньо визначеним [4].
Конфлікт інтересів. Автор заявляє про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці даної статті.

/34.jpg)
/35.jpg)