Туберозний склероз (ТС) — це рідкісне спадкове захворювання з групи факоматозів, вперше згадане у 1880 році французьким неврологом Désiré-Magloire Bourneville, який описав клінічні прояви в пацієнта з ураженням головного мозку, використавши термін «склерозуючі тубери». ТС може проявлятися широким спектром клінічних ознак, у тому числі уражати шкірні покриви, мозок, серце, нирки, а також інші внутрішні органи, і значно погіршує якість життя пацієнтів і скорочує його тривалість за умови відсутності адекватного лікування. Також є характерною значна гетерогенність симптомів ТС у різних органах — перебіг може значно відрізнятися навіть у близькоспоріднених родичів [2].
Частота захворювання, за різними джерелами, може коливатися від 1 : 5800 до 1 : 10 000 випадків [1, 3].
Причиною ТС вважається виникнення гетерозиготної мутації в генах TSC1 чи TSC2 внаслідок мутації de novo або успадкування від одного з батьків, які також мають дану мутацію. Дані гени відповідають за синтез специфічних білків-регуляторів — гамартину і туберину, які в комплексі можуть інгібувати проходження сигналу по сигнальному шляху mTOR, що регулює базові аспекти поведінки клітини, такі як ріст (збільшення маси) і проліферація (поділ), залежно від її енергетичного статусу, наявності/браку амінокислот, сигналів від факторів росту та інших зовнішніх і внутрішніх факторів [6, 10].
Вважається, що розвиток туберів/гамартом потребує двох мутацій, одна із них у першій алелі є успадкованою, а інша виникає як соматична мутація в іншій алелі вже після народження. Мутація в гені TSC2 асоційована з тяжчим перебігом порівняно з TSC1 [7].
Основні клінічні прояви ТС:
— ураження центральної нервової системи:
- субепіндимальна гігантоклітинна астроцитома;
- кортикальні тубери;
- субепіндимальні вузлики;
- лінії міграції білої речовини головного мозку;
— ураження внутрішніх органів, у тому числі серця (рабдоміоми), нирок (ангіоміоліпоми, множинні ниркові кісти), легень (лімфангіолейоміоматоз), зорового апарату (множинні гамартоми сітківки), інших органів і систем (нениркові гамартоми);
— ураження шкірних покривів та їх похідних (ділянки депігментації шкіри, точкові ураження шкіри за типом конфетті, ангіофіброми обличчя, фіброзні бляшки голови, ділянки «шагреневої шкіри», точкові ушкодження емалі зубів, навколонігтьові фіброми, інтраоральні фіброми).
Ушкодження шкіри та її похідних спостерігаються практично в 100 % хворих із підтвердженим діагнозом ТС, найчастіше відзначається депігментація шкіри. Жодне з них не становить загрози здоров’ю чи життю пацієнта, проте можуть призводити до косметичних дефектів шкіри і погіршувати якість життя хворого.
Основними неврологічними проявами ТС є епілептичні напади, що спостерігаються в 70–90 % усіх хворих [4]. Напади зазвичай з’являються в перші три роки життя дитини, маніфестуючи інфантильними і фокальними судомами. В половини усіх хворих відзначаються затримка фізичного і психомовленнєвого розвитку, легка чи помірно виражена розумова відсталість. У 40–50 % пацієнтів можуть спостерігатися симптоми поведінкових розладів за типом РАС. У медичній літературі використовується абревіатура TAND (TSC-Associated Neuropsychiatric Disorder — ТС-асоційований психоневрологічний розлад) — термін, що включає функціональні і клінічні прояви мозкової дисфункції, у тому числі поведінкові, неврологічні, розумові, нейропсихологічні і психосоціальні ускладнення в пацієнтів із ТС [1, 8].
Інші прояви захворювання — рабдоміоми серця становлять собою доброякісні утвори, що, проте, можуть призводити до порушення гемодинаміки і потребувати оперативного втручання.
Ураження з боку нирок — ангіоміоліпоми, кісти нирок — часте ускладнення, що, за деякими даними, є другою за частотою причиною передчасної смерті у хворих на ТС. Основні ускладнення — заочеревинна чи внутрішньониркова кровотеча, як гостра, так і хронічна, рідше зустрічається нирковоклітинна онкоцитома, а також непухлинне ураження нирок — фокальний сегментарний гломерулосклероз. Ці зміни можуть призводити до прогресування ниркової недостатності, однак загалом термінальна хронічна ниркова недостатність виникає не часто [9].
Офтальмологічні ускладнення. Гамартоми сітківки можуть виявлятися у 30–50 відсотків пацієнтів, частіше є двосторонніми і множинними, клінічно себе проявляти можуть вже в новонароджених. У більшості пацієнтів ураження не призводить до серйозних порушень зору.
Діагностика захворювання відбувається згідно з оновленими критеріями 2012 року, ухваленими другою Інтернаціональною колегіальною конференцією з ТС.
До великих критеріїв ТС відносять [3, 6]:
— плями депігментації (3 і більше, діаметром більше 5 мм);
— ангіофіброми (3 і більше) чи фіброзні бляшки голови;
— навколонігтьові фіброми (2 і більше);
— ділянки «шагреневої шкіри»;
— множинні гамартоми сітківки;
— кортикальна дисплазія;
— субепіндимальні вузлики;
— субепіндимальна гігантоклітинна астроцитома;
— рабдоміома серця;
— лімфангіолейоміоматоз;
— ангіоміоліпоми нирок (2 і більше).
До малих критеріїв ТС відносять [3, 6]:
— ураження шкіри за типом конфетті;
— точкові дефекти емалі (2 і більше);
— інтраоральні фіброми (2 і більше);
— ділянки депігментації сітківки;
— множинні кісти нирок;
— нениркові гамартоми.
Згідно з даними критеріями, для установлення діагнозу ТС необхідна наявність однієї значимої мутації в гені TSC1 чи TSC2 або ж наявність двох великих критеріїв чи одного великого і двох малих. Запідозрити діагноз можна за наявності в пацієнта одного великого і двох малих критеріїв [3].
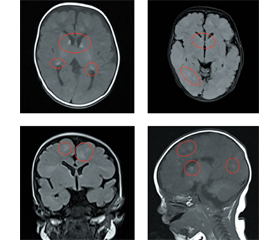
Для установлення діагнозу необхідно провести ретельний огляд пацієнта на наявність зовнішніх фенотипових ознак захворювання згідно з критеріями діагностики, ретельно зібрати сімейний анамнез, а також провести додаткові методи дослідження: ультразвукове дослідження (УЗД) органів черевної порожнини і нирок; ЕхоКГ для виключення рабдоміом; МРТ-обстеження голови для виявлення туберів у головному мозку; за потреби — інші дослідження, що можуть допомогти встановити діагноз, у тому числі і молекулярно-генетичне дослідження для виявлення мутації в генах TSC1 і TSC2.
/61.jpg)
Лікування ТС як захворювання, що може одночасно уражати багато органів і систем, потребує мультидисциплінарного підходу і включає спостереження в таких спеціалістів, як невролог, кардіолог, онколог, дерматолог, нефролог, офтальмолог і генетик. Медикаментозне лікування передбачає прийом протисудомних препаратів відповідно до індивідуальної чутливості пацієнта; також добре себе зарекомендували препарати, дія яких заснована на інгібіції активації комплексу mTOR (препарат сиролімус), надмірна активація якого і лежить в основі патогенезу більшості клінічних проявів ТС [8].
Клінічний випадок. Хотіли б представити вашій увазі випадок спостереження за пацієнткою А. віком 1 рік і 3 місяці, яка спостерігається в Кіровоградській обласній клінічній дитячій лікарні з діагнозом «туберозний склероз».
Відомо, що пробанд від 3-ї вагітності, 3-х пологів. Усі вагітності від одного батька. Від 1-ї вагітності — хлопець, 6,5 року, здоровий, фізичний і психомовленнєвий розвиток згідно з віком, зі слів матері, наявна ділянка депігментації на лівій щоці. Дитина не обстежувалася в генетика. Від 2-ї вагітності — хлопець, 4,5 року, фізичний і психомовленнєвий розвиток згідно з віком, у генетика також не обстежувався.
Пробанд, дівчинка, народилася від 3-ї вагітності, 3-х пологів. Вагітність перебігала на фоні анемії 1 ст., на зберіганні мати не перебувала. На облік у жіночу консультацію стала в 13 тижнів. Вік обох батьків 26 років. У терміні 25 тижнів при проведенні планового УЗД-обстеження були виявлені множинні утвори в порожнині серця, запідозрено рабдоміоми, матір направлена в Київ в ДУ «НПМЦ дитячої кардіології і кардіохірургії», повторно консультована в терміні гестації 34 тижні, висновок: вагітність одним живим плодом терміном 34 тижні вагітності. У плода множинні гіперехогенні утвори в порожнині лівого та правого шлуночків (рабдоміоми?). Імовірність поєднання рабдоміом з патологією головного мозку (ТС) — 90 %.
Вплив шкідливостей: вживання алкоголю, тютюнокуріння, професійні, побутові шкідливості під час вагітності мати заперечує.
Спадковість у батьків пробанда: вроджена вада серця неуточнена в сестри матері пробанда, затримка психомовленнєвого розвитку в племінника матері пробанда, ДЦП — в іншого племінника.
Пологи в перинатальному центрі на 40–41-му тижні вагітності, самостійні. Вага при народженні — 3750 г, довжина тіла — 51 см, за Апгар — 8/9 балів. Дівчинка перебувала сумісно з матір’ю, вигодовування грудне + докорм. Обстежена: ЕхоКГ — множинні рабдоміоми серця, відкрите овальне вікно (ВОВ); УЗД органів черевної порожнини — дифузні зміни печінки без збільшення розмірів, лівосторонній гідронефроз І–ІІ ступеня. У віці 6 діб була переведена в КНП «Обласна клінічна дитяча лікарня Кіровоградської обласної ради» (КНП «ОКДЛ КОР») із матір’ю, де перебувала протягом 12 діб. Консультована кардіологом: множинні рабдоміоми серця, ВОВ; урологом: лівобічний гідронефроз; окулістом: патології не виявлено; неврологом: гіпоксично-ішемічне ураження центральної нервової системи, синдром загальномозкових порушень.
У віці 46 діб дитина повторно була консультована спеціалістами ДУ «НПМЦ дитячої кардіології і кардіохірургії». Установлений діагноз: множинні рабдоміоми в порожнині серця без обструкції кровообігу. ВОВ. Рекомендована консультація спеціалістів обласної лікарні для виключення конкуруючої позасерцевої патології. Проведене МРТ-обстеження голови: МРТ-картина туберозного склерозу (множинні тубери в кортикальних, субкортикальних відділах і глибоких відділах білої речовини обох півкуль головного мозку).
У віці 2,5 місяця дитина повторно була госпіталізована в КНП «ОКДЛ КОР» у супроводі анестезіолога з діагнозом: вроджена вада серця: множинні рабдоміоми серця, судомний синдром, гідронефроз лівої нирки, госпіталізована у відділення анестезіології та інтенсивної терапії. Мати висловлювала скарги на судоми, в’ялість, блювання після їжі, неспокій. Після стабілізації загального стану переведена в кардіоревматологічне відділення. Обстежена: ЕхоКГ — множинні рабдоміоми в порожнині серця без обструкції кровотоку. ЕЕГ: зареєстровані практично постійні білатерально-синхронні комплекси, що розцінено як епіактивність; генетик: туберозний склероз. Дитині було призначено депакін у дозі 60 мг 3 рази на добу тривало, діакарб 1/4 табл. 2 рази на добу, аспаркам 1/3 табл. 3 рази на добу, видано направлення в ДУ «НПМЦ дитячої кардіології і кардіохірургії» та до генетика НДСЛ «Охматдит» із метою встановлення діагнозу, вирішення подальшої тактики ведення й експертних питань.
/62_2.jpg)
Висновок ДУ «НПМЦ дитячої кардіології і кардіохірургії»: множинні рабдоміоми в порожнині правого шлуночка з невеликою обструкцією кровотоку в легеневу артерію, рекомендоване спостереження за місцем проживання. Висновок НДСЛ «Охматдит», спеціалізованого медико-генетичного центру: туберозний склероз, автосомно-домінантний тип успадкування, нова мутація. Рекомендоване оформлення соціальної допомоги строком до 18 років.
У віці 9 місяців дитина повторно надійшла до КНП «ОКДЛ КОР» зі скаргами на судоми, підвищення температури тіла, госпіталізована в інфекційне відділення. Обстежена спеціалістами: невролог: ВАР ГМ — факоматоз, ТС, симптоматична епілепсія, часті міоклонічні напади. Кардіолог: ВАР ГМ — факоматоз, ТС, множинні рабдоміоми в порожнині вихідного тракту правого шлуночка; окуліст: незначна ангіопатія ОО; ЕЕГ: поодинокі специфічні ЕЕГ-феномени. Скореговано протисудомне лікування, призначено депакін у дозі 70 мг 3 рази на добу, 1 місяць, із контролем ЕЕГ після лікування для корекції дозування; сабрил 500 мг — 1/4 табл. 2 рази на добу, тривало.
Об’єктивно: на момент першого огляду лікарем-генетиком дитини віком 2 місяці серед очевидних фенотипових ознак відзначалися такі: ангіоми блідо-червоного і червоного кольорів, розташовані в ділянці верхнього плечового поясу, а також на правій сідниці, голубуваті склери, випинання лобних горбів, ділянки депігментації не візуалізувалися. У віці 8 місяців: численні ділянки депігментації діаметром 1 см і більше на тулубі, в паху; одна з гемангіом, описана вище; на правій руці поява ущільнення під незміненою шкірою. У віці 11 місяців з’явилося ущільнення на шиї праворуч під незміненою шкірою, неболюче.
На момент останнього огляду під час планової госпіталізації в неврологічному відділенні у віці 1 року і 3 місяців: вага 12 кг (85–97-й перцентиль за вагою). Дитина самостійно сидить, стоїть, активно повзає, пробує ходити. Вимовляє окремі слова, активно лепече. Судом на фоні прийому препаратів не відзначалося. Фенотипово: високий лоб, ангіома в ділянці грудної клітки праворуч, численні ділянки депігментації на шкірі, їх кількість порівняно з попереднім оглядом збільшилася, ущільнення на шкірі. Отримує лікування: депакін 75 мг 2 рази на добу тривало, сабрил 500 мг — 1/2 табл. 2 рази на добу тривало.
Дані обстежень:
— УЗД органів черевної порожнини: структурних змін органів черевної порожнини не виявлено.
— УЗД нирок: розширення верхньої чашки праворуч.
— ЕЕГ: позитивна динаміка у вигляді часткового регресу епіактивності.
— ЕхоКГ: множинні рабдоміоми вихідного тракту правого шлуночка з невеликою обструкцією кровотоку в легеневу артерію.
Висновки
Таким чином, правильна і своєчасна діагностика синдрому туберозного склерозу в дітей і дорослих пацієнтів може становити значний виклик для лікарів не тільки первинної ланки, а й лікарів-спеціалістів різних профілів, оскільки ТС є полісистемним захворюванням, і перед тим як буде встановлено діагноз, дитина може бути проконсультована декількома спеціалістами. У даної пацієнтки діагноз був запідозрений ще до народження завдяки проведеному УЗ-скринінгу, а після народження — підтверджений виявленими іншими великими критеріями — наявністю туберів у головному мозку й уражень шкіри. Проте не завжди рабдоміоми діагностуються вчасно, і першими клінічними проявами будуть зміни шкірних покривів, що можуть бути не розпізнані лікарями-педіатрами і призводити до відтермінування призначення необхідного лікування, погіршення якості життя та зниження його тривалості.
Подібний клінічний випадок, так званий «складний пацієнт», на наш погляд, має спостерігатися мультидисциплінарною командою лікарів-фахівців, що сприяє своєчасному уточненню діагнозу, призначенню додаткових обстежень, правильному лікуванню та нагляду за дитиною.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці даної статті.
Отримано/Received 21.08.2021
Рецензовано/Revised 06.09.2021
Прийнято до друку/Accepted 13.09.2021

/61.jpg)
/62.jpg)
/62_2.jpg)