
Журнал «Здоровье ребенка» Том 18, №3, 2023
Вернуться к номеру
Атиповий гемолітико-уремічний синдром: клінічний випадок у дитячому віці
Авторы: K.K. Hodiatska (1), T.K. Mavropulo (1), T.A. Bordii (1), S.V. Alifanova (1), V.F. Doroshenko (2), L.M. Cherhinets (2)
(1) — Dnipro State Medical University, Dnipro, Ukraine
(2) — City Children’s Clinical Hospital 6, Dnipro, Ukraine
Рубрики: Педиатрия/Неонатология
Разделы: Справочник специалиста
Версия для печати
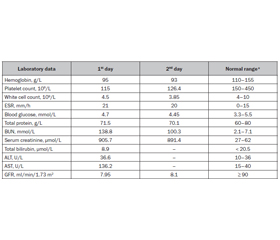
Атиповий гемолітико-уремічний синдром (aГУС) є надзвичайно рідкісним, але небезпечним для життя захворюванням у дітей через те, що може спричинити гостре пошкодження нирок. Пацієнти з aГУС мають ризик розвитку повторних епізодів. Отже, у цій статті ми представляємо випадок рецидиву аГУС у хлопчика 9 років. Дитина надійшла у відділення невідкладної допомоги на п’ятий день хвороби з основними скаргами на одутлість обличчя та зменшення виділення сечі. З анамнезу відомо, що в пацієнта розвинувся другий епізод aГУС через шість років після повного одужання від першого епізоду. Клінічні прояви обох епізодів аГУС були спровоковані інфекцією дихальних шляхів, гастроінтестинальні симптоми не відмічалися. Результати діагностичних досліджень, проведених під час першого епізоду аГУС, наступні: аналіз калу на Escherichia coli й токсини Shiga був негативним; дослідження комплементу не виявили порушень; активність ADAMTS13 і рівень антитіл до фактора Н комплементу були в нормі. Результати ультразвукового дослідження та біопсії нирок відповідали діагнозу. Сімейний анамнез характеризувався діагнозом аГУС у молодшого брата пацієнта, що було підтверджено молекулярно-генетичним тестуванням, зокрема виявлено патогенний варіант у гені CD46/MCP (мембранний кофакторний білок) у гетерозиготному стані. При фізикальному обстеженні зафіксовано блідість, набряк обличчя, помірну артеріальну гіпертензію, олігурію. Лабораторно виявлено гемолітичну анемію, тромбоцитопенію, значну азотемію, різке зниження клубочкової фільтрації, високий рівень аспартатамінотрансферази, незначний електролітний дисбаланс, протеїнурію. Підтримуюче лікування включало інфузійну терапію, використання свіжозамороженої плазми, фуросеміду й дексаметазону. Дитині розпочато гострий гемодіаліз унаслідок тяжкого гострого пошкодження нирок. Висновки. Рецидив аГУС характеризується тяжкою нирковою недостатністю, що потребує гострого гемодіалізу. Вірусні інфекції є потенційними тригерами аГУС. Повторний перебіг захворювання та позитивний сімейний анамнез вказують на важливість генетичного скринінгу, оскільки слід розглядати наявність сімейного аГУС.
Background. Atypical hemolytic uremic syndrome (aHUS) is an extremely rare but life-threatening disorder in children since it may cause acute kidney injury. Patients with aHUS are at risk of recurrence. Hence, in this paper, we present a case of a 9-year-old boy with aHUS relapse. The child was admitted to the emergency department on the fifth day of illness with main complaints of facial puffiness and decreased urine output. Based on the medical history, the patient developed the second episode of aHUS after 6 years of complete recovery from the first episode. There was no preceding diarrheal illness, instead, the clinical manifestation of both aHUS episodes was triggered by a respiratory tract infection. The results of diagnostic studies performed during the first episode of aHUS were as follows: stool tests for Escherichia coli and Shiga toxins were negative; a complement assay showed no abnormalities; ADAMTS13 activity and anti-complement factor H antibodies were normal. The results of the kidney ultrasonography and biopsy were consistent with the diagnosis. Family history was remarkable for aHUS in a younger sibling confirmed by molecular genetic testing, in particular, a pathogenic variant in the CD46/MCP (membrane cofactor protein) gene in the heterozygous state has been identified. Physical examination revealed paleness, facial swelling, moderate hypertension, and oliguria. Laboratory findings demonstrated hemolytic anemia, thrombocytopenia, significant azotemia, a severe reduction in the glomerular filtration rate, a high level of aspartate aminotransferase, insignificant electrolyte imbalance, and proteinuria. Supportive treatment included fluid and electrolyte management, fresh frozen plasma, furosemide, and dexamethasone. The child commenced acute hemodialysis due to severe acute kidney injury. Conclusions. A recurrence of aHUS is characterized by severe renal failure requiring acute hemodialysis. Viral infections are potential triggers of aHUS. A relapsing course of the disease and a family history of aHUS indicate the importance of genetic screening, as familial aHUS should be considered.
атиповий гемолітико-уремічний синдром; діти; рецидив
atypical hemolytic uremic syndrome; children; relapse
Для ознакомления с полным содержанием статьи необходимо оформить подписку на журнал.
- Yan K., Desai K., Gullapalli L., Druyts E., Balijepalli C. Epidemiology of atypical hemolytic uremic syndrome: a systematic literature review. Clin. Epidemiol. 2020 Mar 12. 12. 295-305. doi: 10.2147/CLEP.S245642.
- Noris M., Bresin E., Mele C., Remuzzi G. Genetic atypical hemolytic-uremic syndrome. 2007 Nov 16. In: Adam M.P., Mirzaa G.M., Pagon R.A. et al., eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2023.
- Gülhan B., Özaltın F. Hemolytic uremic syndrome in children. Turk. Arch. Pediatr. 2021 Sep. 56(5). 415-422. doi: 10.5152/TurkArchPediatr.2021.21128.
- Gurevich E., Landau D. Pharmacological management of atypical hemolytic uremic syndrome in pediatric patients: current and future. Paediatr. Drugs. 2023 Mar. 25(2). 193-202. doi: 10.1007/s40272-022-00555-6.
- Afshar-Kharghan V. Atypical hemolytic uremic syndrome. Hematology Am. Soc. Hematol. Educ. Program. 2016 Dec 2. 2016(1). 217-225. doi: 10.1182/asheducation-2016.1.217.
- Mano L., Francisco T., Gaspar J., Pereira G., Santos R., Abran–ches M. Influenza B-associated atypical hemolytic uremic syndrome. Port. J. Nephrol. Hypert. 2022. 36(2). 80-83. doi: 10.32932/pjnh.2022.06.180.
- Raina R., Vijayvargiya N., Khooblall A. et al. Pediatric atypical hemolytic uremic syndrome advances. Cells. 2021 Dec 18. 10(12). 3580. doi: 10.3390/cells10123580.
- Bagga A., Khandelwal P., Mishra K. et al. Hemolytic uremic syndrome in a developing country: Consensus guidelines. Pediatr. Nephrol. 2019 Aug. 34(8). 1465-1482. doi: 10.1007/s00467-019-04233-7.
- Angioi A., Fervenza F.C., Sethi S. et al. Diagnosis of complement alternative pathway disorders. Kidney Int. 2016 Feb. 89(2). 278-288. doi: 10.1016/j.kint.2015.12.003.
- Bu F., Borsa N., Gianluigi A., Smith R.J. Familial atypical hemolytic uremic syndrome: a review of its genetic and clinical aspects. Clin. Dev. Immunol. 2012. 2012. 370426. doi: 10.1155/2012/370426.
- Joseph C., Gattineni J. Complement disorders and hemolytic uremic syndrome. Curr. Opin. Pediatr. 2013 Apr. 25(2). 209-215. doi: 10.1097/MOP.0b013e32835df48a.
- Avila Bernabeu A.I., Cavero Escribano T., Cao Vilarino M. Aty–pical hemolytic uremic syndrome: new challenges in the complement blo–ckage era. Nephron. 2020. 144(11). 537-549. doi: 10.1159/000508920.
- Le Clech A., Simon-Tillaux N., Provôt F. et al. Atypical and se–condary hemolytic uremic syndromes have a distinct presentation and no common genetic risk factors. Kidney Int. 2019 Jun. 95(6). 1443-1452. doi: 10.1016/j.kint.2019.01.023.
- Loirat C., Fakhouri F., Ariceta G. et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2016 Jan. 31(1). 15-39. doi: 10.1007/s00467-015-3076-8.
- Kobbe R., Schild R., Christner M., Oh J., Loos S., Kemper M.J. Case report — atypical hemolytic uremic syndrome triggered by influenza B. BMC Nephrol. 2017 Mar 20. 18(1). 96. doi: 10.1186/s12882-017-0512-y.
- Khandelwal P., Krishnasamy S., Govindarajan S. et al. Anti-factor H antibody associated hemolytic uremic syndrome following SARS-CoV-2 infection. Pediatr. Nephrol. 2022 Sep. 37(9). 2151-2156. doi: 10.1007/s00467-021-05390-4.
- Sridharan M., Go R.S., Abraham R.S. et al. Diagnostic utility of complement serology for atypical hemolytic uremic syndrome. Mayo Clin. Proc. 2018 Oct. 93(10). 1351-1362. doi: 10.1016/j.mayocp.2018.07.008.
- Raina R., Mangat G., Hong G. et al. Anti-factor H antibody and its role in atypical hemolytic uremic syndrome. Front. Immunol. 2022 Aug 23. 13. 931210. doi: 10.3389/fimmu.2022.931210.
- Waters A.M., Licht C. aHUS caused by complement dysregulation: new therapies on the horizon. Pediatr. Nephrol. 2011 Jan. 26(1). 41-57. doi: 10.1007/s00467-010-1556-4.
- Ardissino G., Longhi S., Porcaro L. et al. Risk of atypical HUS among family members of patients carrying complement regulatory gene abnormality. Kidney Int. Rep. 2021 Mar 25. 6(6). 1614-1621. doi: 10.1016/j.ekir.2021.03.885.
- Nester C.M., Barbour T., de Cordoba S.R. et al. Atypical aHUS: state of the art. Mol. Immunol. 2015 Sep. 67(1). 31-42. doi: 10.1016/j.molimm.2015.03.246.
- Diamante Chiodini B., Davin J.C., Corazza F. et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics. 2014 Jun. 133(6). e1764-1768. doi: 10.1542/peds.2013-1594.