
Журнал «Боль. Суставы. Позвоночник» Том 13, №3, 2023
Вернуться к номеру
Дерматоспараксійний тип синдрому Елерса — Данло: клінічний випадок
Авторы: Балацька Н.І.(1), Строй О.А. (1), Гриневич І.В. (2), Гусинін П.В. (3), Медведєва О.П. (4), Кирильчук К.Ю. (4)
(1) — Національний медичний університет імені О.О. Богомольця, м. Київ, Україна
(2) — ВПНЗ «Київський медичний університет», м. Київ, Україна
(3) — ДУ «Національний інститут серцево-судинної хірургії імені М.М. Амосова НАМН України», м. Київ, Україна
(4) — Консультативно-діагностична поліклініка НДСЛ «Охматдит» МОЗ України, м. Київ, Україна
Рубрики: Ревматология, Травматология и ортопедия
Разделы: Справочник специалиста
Версия для печати
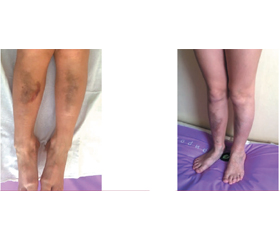
Актуальність. Дерматоспараксійний тип синдрому Елерса — Данло (EDSDERMS, VIIC, dEDS) — надзвичайно рідкісне орфанне захворювання. На сьогодні в усьому світі відомо про 15 пацієнтів із цим типом синдрому Елерса — Данло. Мета: поширення даних про орфанне захворювання сполучної тканини — dEDS — на прикладі пацієнта із сім’ї, у якій у батька і двох синів є однотипні ураження. Матеріали та методи. Ми повідомляємо про 6-річного пацієнта, його батька і сиблінга з клінічними симптомами dEDS, який ми діагностували на основі таких великих діагностичних критеріїв: надзвичайна крихкість шкіри, характерні черепно-лицеві риси, наявність поверхневих травм шкіри, зморшкуватість шкіри долонь, особливо при тривалому перебуванні їх у воді, схильність до екхімозів з ризиком підшкірних гематом і кровотеч. Наявні такі малі діагностичні критерії: атрофічні рубці, порушення рефракції (міопія), дисплазія ясен гінгівального краю, гіпермобільність суглобів пальців і колінних суглобів. Була отримана інформована згода батьків пацієнта на обстеження, публікацію результатів досліджень і клінічних фотографій. Результати. Після лабораторно-інструментальних досліджень і консультацій суміжних фахівців (гематолог, генетик, кардіолог, педіатр, ортопед, дерматолог), виключення таких захворювань, як синдром Марфана, MASS-фенотип, вроджені коагулопатії та тромбоцитопатії, гемофілія, ізольовані судинні патології, а також інші типи синдрому Елерса — Данло, ми ідентифікували у пацієнта dEDS лише клінічно. Генетичне дослідження не виявило патологічних мутацій, екзонних делецій/дуплікацій. Негативний результат генетичного тестування при dEDS обумовлений тим, що певні типи мутацій (наприклад, глибокі інтронні мутації) не завжди можна виявити за допомогою стандартних діагностичних генетичних методів. Висновки. Можливе встановлення діагнозу за клінічними симптомами, але розширений пошук мутацій обов’язковий для всієї сім’ї.
Background. The dermatosparaxial type of Ehlers-Danlos syndrome (EDSDERMS, VIIC, dEDS) is an extremely rare disorder. To date, 15 patients with this type of Ehlers-Danlos syndrome are known worldwide. The purpose was to improve knowledge and spread data about the orphan connective tissue disease — dEDS — on the example of a patient from a family in which the father and two sons have the same type of lesions. Materials and methods. We report a 6-year-old patient as well as his father and sibling with clinical symptoms of the dEDS, which we diagnosed based on the main diagnostic criteria: extreme skin fragility, craniofacial features, superficial skin trauma, wrinkling of the palms, especially when they are exposed to water for a long time, and a tendency to ecchymosis with a risk of subcutaneous hematomas and bleeding. Secondary diagnostic criteria were represented by atrophic scars, refractive errors (myopia), gingival marginal dysplasia, hypermobility of the finger and knee joints. The informed consent of the patient’s parents was obtained for the examination and publication of information, and the parents agreed to the publication of clinical photographs. Results. After laboratory and instrumental studies and consultations with related specialists (hematologist, geneticist, cardiologist, pediatrician, orthopedist, dermatologist), we excluded such diseases as Marfan syndrome, MASS-phenotype, congenital coagulopathies and thrombocytopathies, hemophilia, isolated vascular pathologies, as well as other types of Ehlers-Danlos syndrome and identified dEDS in the patient only clinically. Genetic testing did not reveal any pathological mutations or exonic deletions/duplications. The negative result of genetic testing in dEDS is due to the fact that certain types of mutations (e.g., deep intronic mutations) cannot always be detected by standard diagnostic genetic methods. Conclusions. It is possible to establish a diagnosis based on clinical symptoms, but an extended mutation search is mandatory for the entire family.
орфанне захворювання; синдром Елерса — Данло; дерматоспараксійний тип; клінічний випадок; геморагічний синдром
orphan disease; Ehlers-Danlos syndrome; dermatosparaxis type; hemorrhagic syndrome; case report
Вступ
Клінічний випадок
/105.jpg)
/106.jpg)
/106_2.jpg)
Обговорення
Висновки
- Beighton P., De Paepe A., Steinmann B et al. Ehlers-Danlos syndromes: revised nosology, Villefranche. 1997. Am. J. Med. Genet. 1998. 77(1). 31-7. doi: https://doi.org/10.1002/(SICI)1096-8628(19980428)77:1<31::AID-AJMG8>3.0.CO;2-O.
- Royce P.M., Steinmann В. Connective tissue and its heritable disorders: molecular, genetic and medical aspects. 2nd ed. New York: Wiley-Liss, Inc., 2002. 1190 р.
- Ritelli M., Colombi M. Molecular Genetics and Pathogenesis of Ehlers-Danlos Syndrome and Related Connective Tissue Disorders. Genes. 2020. 11(5). 547. DOI.10.3390/genes11050547.
- Malfait F., Castori M., Francomano C.A. et al. The Ehlers-Danlos syndromes. Nat. Rev. Dis. Primers. 2020 Jul 30. 6(1). 64. doi: 10.1038/s41572-020-0194-9. PMID: 32732924.
- Hein L.C., DeGregory C.B., Umari F. Ehlers-Danlos Syndrome: It’s Not Your Normal Hoofbeats. J. Nurse Pract. 2019. 15. 277-281. doi: 10.1016/j.nurpra.2019.01.006.
- Song B., Yeh P., Harrell J. Systemic manifestations of Ehlers-Danlos syndrome. Proceedings. Proc. (Bayl. Univ. Med. Cent.). 2021. 34(1). 49-53. doi: 10.1080/08998280.2020.1805714.
- Gensemer C., Burks R., Kautz S., Judge D.P., Lavallee M., Norris R.A. Hypermobile Ehlers-Danlos syndromes: Complex phenotypes, challenging diagnoses, and poorly understood causes. Dev. Dyn. 2021. 250(3). 318-344. doi: 10.1002/dvdy.220.
- McGillis L., Mittal N., Santa Mina D. et al. Utilization of the 2017 diagnostic criteria for hEDS by the Toronto GoodHope Ehlers-Danlos syndrome clinic: A retrospective review. Am. J. Med. Genet. A. 2020 Mar. 182(3). 484-492. doi: 10.1002/ajmg.a.61459.
- Van Damme T., Colman M., Syx D., Malfait F. The Ehlers-Danlos Syndromes against the Backdrop of Inborn Errors of Metabolism. Genes (Basel). 2022. 13(2). 265. doi: 10.3390/genes13020265.
- Riley B. The Many Facets of Hypermobile Ehlers-Danlos Syndrome. J. Am. Osteopath. Assoc. 2020 Jan 1. 120(1). 30-32. doi: 10.7556/jaoa.2020.012.
- Gensemer C., Burks R., Kautz S. Hypermobile et al. Ehlers-Danlos syndromes: Complex phenotypes, challen–ging diagnoses, and poorly understood causes. Dev. Dyn. 2021 Mar. 250(3). 318-344. doi: 10.1002/dvdy.220.
- Malfait F., Francomano C., Byers P. et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C. Semin. Med. Genet. 2017. 175(1). 8-26. doi: 10.1002/ajmg.c.31552.
- Lapiere C.M., Lenaers A., Kohn L.D. Procollagen peptidase: an enzyme excising the coordination peptides of collagen. Proc. Nat. Acad. Sci. USA. 1971. 68. 3054-3058. doi: 10.1073/pnas.68.12.3054.
- Lichtenstein J.R., Martin G.R., Kohn L.D., Byers P.H., McKusick V.A. Defect in conversion of procollagen to collagen in a form of Ehlers-Danlos syndrome. Science. 1973. 182. 298-299. doi: 10.1126/science.182.4109.298.
- Smith L.T., Wertelecki W., Milstone L.M. Human dermatosparaxis: a form of Ehlers-Danlos syndrome that results from failure to remove the amino-terminal propeptide of type I procollagen. Am. J. Hum. Genet. 1992. 51(2). 235-44. PMCID: PMC1682688.
- Brady A.F., Demirdas S., Fournel-Gigleux S. et al. The Ehlers-Danlos syndromes, rare types. Am. J. Med. Genet. C. Semin. Med. Genet. 2017. 175(1). 70-115. doi: 10.1002/ajmg.c.31550.
- Van Damme T., Colige A., Syx D. et al. Expanding the clinical and mutational spectrum of the Ehlers-Danlos syndrome, dermatosparaxis type. Genet. Med. 2016. 18. 882-891. doi: 10.1038/gim.2015.188.
- Olubajo F., Kaliaperumal C., Choudhari K.A. Vascular Ehlers-Danlos Syndrome: Literature review and surgical management of intracranial vascular complications. Clin. Neurol. Neurosurg. 2020 Jun. 193. 105775. doi: 10.1016/j.clineuro.2020.105775.
- Malfait F. Vascular aspects of the Ehlers-Danlos Syndromes. Matrix Biol. 2018 Oct. 71-72. 380-395. doi: 10.1016/j.matbio.2018.04.013. Epub 2018 Apr 27.
- Colige A., Nuytinck L., Hausser I. et al. Novel Types of Mutation Responsible for the Dermatosparactic Type of Ehlers-Danlos Syndrome (Type VIIC) and Common Polymorphisms in the ADAMTS2 Gene. Journal of Investigative Dermatology. 2004. 123(4). 656-663. doi: 10.1111/j.0022-202X.2004.23406.x.