
Журнал «Здоровье ребенка» 2 (61) 2015
Вернуться к номеру
Патология сердечно-сосудистой системы у детей с синдромом Шерешевского — Тернера
Авторы: Абатуров А.Е., Петренко Л.Л., Агафонова Е.А. — ГУ «Днепропетровская медицинская академия» МЗ Украины; Абатурова Н.И. — КУ «Днепропетровская областная детская клиническая больница» ДОС
Рубрики: Педиатрия/Неонатология
Разделы: Клинические исследования
Версия для печати
Статья посвящена изучению структуры патологии сердечно-сосудистой системы у детей с синдромом Шерешевского — Тернера. Результаты работы основаны на данных обследования 42 больных с синдромом Шерешевского — Тернера в возрасте от 1,5 до 17 лет. Показано, что у 80,9 % больных с синдромом Шерешевского — Тернера отмечаются патологические изменения со стороны сердечно-сосудистой системы разной степени выраженности. Наиболее часто встречаемыми врожденными пороками сердца при синдроме Шерешевского — Тернера являются коарктация аорты, двустворчатый аортальный клапан и дефект межпредсердной перегородки. Синдром Шерешевского — Тернера в 35,6 % случаев ассоциирован с нарушениями электрической работы сердца, в частности с синдромом удлиненного интервала QT. Атерогенные изменения липидного профиля у больных с синдромом Шерешевского — Тернера в 21,4 % случаев наблюдаются с детства.
Стаття присвячена вивченню структури патології серцево-судинної системи в дітей із синдромом Шерешевського — Тернера. Результати роботи засновані на даних обстеження 42 хворих із синдромом Шерешевського — Тернера віком від 1,5 до 17 років. Показано, що у 80,9 % хворих із синдромом Шерешевського — Тернера визначаються патологічні зміни з боку серцево-судинної системи різного ступеня вираженості. Уроджені вади серця, які найбільш часто зустрічаються при синдромі Шерешевського — Тернера, це коарктація аорти, двостулковий аортальний клапан і дефект міжпередсердної перегородки. Синдром Шерешевського — Тернера в 35,6 % випадків асоційований із порушеннями електричної роботи серця, зокрема з синдромом подовженого інтервалу QT. Атерогенні зміни ліпідного профілю у хворих із синдромом Шерешевського — Тернера в 21,4 % випадків спостерігаються з дитинства.
The paper studies the structure of the pathology of the cardiovascular system in children with Turner syndrome. The results of the survey are based on the findings from 42 patients with Turner syndrome, aged 1.5 to 17 years. It is shown that in 80.9 % of patients with Turner syndrome we have marked pathological changes in the cardiovascular system of varying severity. The most common congenital heart diseases in Turner syndrome are aortic coarctation, bicuspid aortic valve and atrial septal defect. Turner syndrome in 35.6 % of cases is associated with impaired electric work of the heart, in particular, with the long QT syndrome. Atherogenic changes in the lipid profile of patients with Turner syndrome in 21.4 % of cases occur since childhood.
синдром Шерешевского — Тернера, дети, сердечно-сосудистая система.
синдром Шерешевського — Тернера, діти, серцево-судинна система.
Turner syndrome, children, cardiovascular system.
Статья опубликована на с. 23-28
Введение
Синдром Шерешевскогo — Тернера (СШТ) (синдром Ульриха — Тернера, ХО-синдром, моносомия X, дисгенезия гонад) — это хромосомное заболевание, в основе которого лежит частичная или полная потеря одной из двух Х-хромосом в большинстве или во всех клетках организма и которое характеризуется промежуточным полом. Синдром Шерешевского — Тернера встречается с частотой 1 : 2500 — 1 : 3000 живых новорожденных фенотипических девочек и является самой распространенной причиной первичного гипогонадизма [36].
Впервые данный синдром был представлен в 1925 году русским врачом Николаем Адольфовичем Шерешевским, который описал 25-летнюю пациентку с женским фенотипом, субнанизмом, гипогонадизмом, короткой шеей с крыловидными складками, микрогнатией, высоким небом, гипертелоризмом сосков [4]. В 1930 году немецким детским врачом Отто Ульрихом (Otto Ullrich) был представлен пациент со сходной клинической симптоматикой, и в 1938 году американский эндокринолог Генри Тернер (Henry H. Turner) также описал данный синдром. Потому в англоязычной литературе данный синдром получил название «синдром Тернера», в немецкой — «синдром Ульриха — Тернера», а в отечественной — «синдром Шерешевского — Тернера». Синдром Шерешевского — Тернера как хромосомная патология ХО впервые был определен С.Е. Фордом (С.Е. Ford) в 1959 году [3, 26].
В основе анеуплоидии, наблюдаемой при СШТ, лежит нарушение расхождения хромосом. Изменения кариотипа у больных с СШТ в большинстве случаев характеризуются:
1) отсутствием одной X-хромосомы (45,X), в том числе мозаичностью (50–75 %), 45,X/46,XX (10–15 %), 45,X/46,XY (2–6 %), 45,X/46,X,i (Xq), 45,X/46,Xdel (Xp), 45,X/46,XX/47, XXX;
2) полной или частичной делецией короткого плеча Х-хромосомы (46,Xdel (Xp)) (30 %);
3) формированием изохромосомы Xq (46,XiXq, с двумя одинаковыми плечами Xq и делецией Xp) (5 %), кольцевой хромосомой Х (46, X,r (X)) [14].
Варианты кариотипов у пациентов с синдромом Шерешевского — Тернера [1]:
— 45,X
— 45,X/46,XX
— 45,X/47,XXX
— 45,X/46,XX/47,XXX
— 45,X/47,XXX/48,XXXX
— 45,X/46,XX /47,XXX/48,XXXX
— 45,X/46,X, psudic (Y) (q11.2)
— 46,X, +mar (derX)
— 45,X/46,X, +mar (derX)
— 45,X/46,X, del (X) (p)
— 45,X/46,X, del (X) (q)
— 45,X/46,X, del (X) (q23)
— 46,X, del (X) (p)
— 46,X,r (X)
— 45,X/46,X,r (X)
— 45,X/46,X,r (X) (p22.3q28)
— 45,X/46,X,r (X) (p21q25)/46,XX
— mos45,X/46,X,terrea (X;X) (p21.3;p22.3)
— mos 45,X,del (X) (p22.1) [35]/45,X [19]/46,X,r (X) (p22.1q28) [6]
— 46,X, del (X) (pter- > q21.1::p11.4- > pter)/45,X
— 46,X.i (X) (q10)
— 45,X/46,X.i (X) (q10)
— 45,X/46,X.i (X) (q10)/46,XX
— 45,X/47,XX.i (X) (q10)/46,XX
— 46,X.i (X) (p10)
— 45,X/46,X.i (X) (p10)
— 45,X/46,X.i (X) (p10) /46,XX
— 45,X/46,X.idic (Y)
— 45,X/46,X.idic (Yp)
— 45,X/46,XY
— 45,X/47,XYY
— 45,X/47,XYY/46,XY
— 45,X/46,X,del (Y) (q)
— 45,X/46,X,dic (Y)
Установлено, что моносомия X-хромосомы проявляется в 1–2 % случаев от всех зачатий у человека. У больных с X-хромосомным мозаицизмом возможно наличие клеточных клонов, содержащих Y-хромосому (45,X/46,XY), а также вероятны варианты транслокации между Х-хромосомой и аутосомами. У больных с СШТ чаще наблюдается отсутствие отцовской Х-хромосомы. Синдром Тернера не связан с возрастом матери, вероятно, его развитие обусловлено нестабильностью Y-хромосомы. Считается, что потеря материнской Х-хромосомы приводит к развитию более тяжелых пороков развития внутренних органов. Плоды с кариотипом 45,Х спонтанно абортируются в 98 % случаев. X-хромосомный мозаицизм, обусловленный нарушением дробления зиготы, может сопровождаться наличием клонов клеток, содержащих две Х-хромосомы (45,Х/46,ХХ), Х- и Y-хромосомы (45,X/46,XY), либо клоны с полисомией Х-хромосомы (например, 45,Х/47,ХХХ) [4, 7, 40, 41].
Ernest B. Hook, Dorothy Warburton считают, что все живорожденные с кариотипом 45,Х являются скрытыми, «загадочными» мозаиками, около 60 % из них — мозаики по Х-хромосоме, а 40 % — по Y-хромосоме [21].
Повторный риск рождения ребенка с СШТ невелик, за исключением тех случаев, когда у одного или обоих родителей имеется наследуемая Х-аутосомная транслокация или когда мать несет клон клеток 45,Х [2, 4].
Разнообразие нарушений кариотипа предопределяет фенотипический полиморфизм СШТ. Так, низкий рост встречается у 94,5 % больных СШТ, гипогонадизм — у 95–99 %, аномалии скелета и кардиоваскулярной системы— у 40 %, аномалии почек — у 33–70 % [2]. В настоящее время показано, что отсутствие критического участка X-хромосомы в регионе короткого плеча p11.2–22.1, где картируются гены UBE1; USP9X; BMP15, приводит к проявлению таких признаков, как низкорослость, черепно-лицевые аномалии, гипогонадизм, аутоиммунные заболевания щитовидной железы [1]. Делеция в теломерном псевдоаутосомном участке хромосомы Х pter 22.32, где картируется гомеобокс-содержащий ген SHOX (short stature homeobox), сопровождается задержкой развития продольной длины тела и такими фенотипическими дисморфиями, как вальгусная деформация нижних конечностей, олигодактилия, микрогнатия, «готическое небо» [12].
Тяжесть проявлений СШТ, как правило, коррелирует с процентом клеток, которые несут одну Х-хромосому [33]. При хромосомном мозаицизме 45,Х/46,XX; 45,X/46,XX/47,XXX; 45,X/47,XXX заболевание характеризуется более мягкой клинической картиной. Фенотип пациентов с кариотипом 45,Х/46,ХY варьирует от почти нормального мужского до почти нормального женского с некоторыми признаками СШТ. Интересно, что 90 % мозаиков 45,X/46,XY в пренатальный период жизни имеют мужской фенотип, в то время как в постнатальном периоде наиболее часто встречаются пациентки с признаками СШТ [2].
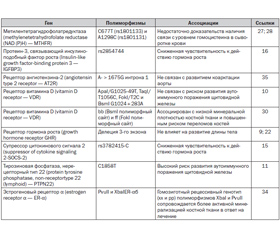
В последнее время показано, что фенотипические проявления СШТ также связаны с однонуклеотидными полиморфизмами (SNP) различных генов (табл. 1).
/19/19.jpg)
Распространенность врожденных пороков сердца у пациентов с СШТ колеблется от 17 до 45 % [13]. Наиболее высокий риск наличия и развития сердечно-сосудистых нарушений характерен для лиц с кариотипом 45,X (OR = 3,50, 95% ДИ 0,99–12,29; р = 0,05) [6].
Сердечно-сосудистые нарушения у больных с СШТ также связаны с риском развития артериальной гипертензии, гиперлипидемии, ожирения и сахарного диабета [8, 32].
Однако, несмотря на высокую частоту встречаемости сердечно-сосудистой патологии при СШТ, данный аспект фенотипического проявления моносомии X-хромосомы у детей представлен в единичных работах. Laura Lucaccioni и соавт. [25] считают необходимыми дальнейшие исследования сердечно-сосудистой системы у больных с СШТ для усовершенствования оценки риска развития неблагоприятных событий и оптимизации ведения пациентов.
Целью исследования явилось изучение структуры частоты встречаемости патологии сердечно-сосудистой системы у детей с СШТ.
Материалы и методы исследования
Под нашим наблюдением находилось 42 пациента с СШТ в возрасте от 1,5 до 17 лет, которые были обследованы в областном эндокринологическом отделении КУ «Городская детская клиническая больница № 1» ДОС г. Днепропетровска и областном кардиоревматологическом отделении КУ «Днепропетровская областная детская клиническая больница» ДОС.
Диагноз СШТ был подтвержден кариотипированием, проведенным в Республиканском генетическом центре г. Кривого Рога. Всем детям были проведены электрокардиография (ЭКГ), эхокардиография (ЭхоКГ). Также проводились исследования уровня инсулиноподобного фактора роста I (ИФР-I), кортизола, фолликулостимулирующего гормона (ФСГ), лютеинизирующего гормона (ЛГ), эстрадиола в сыворотке крови; оценивался характер липидного профиля.
Статистическая обработка полученных результатов проводилась с помощью стандартных методов статистического анализа с использованием программного обеспечения для персонального компьютера Microsoft Excel и Statistica 6.0.
Результаты и обсуждение
У детей с СШТ пороки развития сердечно-сосудистой системы отмечались в 80,9 % случаев. Коарктация аорты была диагностирована у 4 (26,6 %) пациентов с СШТ, у 5 (33,3 %) — двустворчатый аортальный клапан, у 3 (20 %) — дефект межпредсердной перегородки, также было по 1 случаю (6,6 %) открытого артериального протока, сочетания стеноза устья аорты и дефекта межпредсердной перегородки и коарктации аорты в сочетании с двустворчатым клапаном аорты. Четырем пациенткам с целью уточнения диагноза была проведена магнитно-резонансная томография (МРТ) сердца, позволившая выявить аномалии развития верхней полой вены, подключичной артерии, аномальный дренаж легочных вен. Пролапс митрального клапана был диагностирован у 8 больных (23,4 %). Утолщение межпредсердной и межжелудочковой перегородок наблюдалось у 4 пациентов (11,7 %).
Согласно данным других авторов, если в структуре врожденных пороков сердца у детей общей популяции преобладают дефект межжелудочковой перегородки (25 %), дефект межпредсердной перегородки (15 %) и открытый артериальный проток (15 %), то у пациентов с СШТ чаще встречаются коарктация аорты (30 %), бикуспидальный аортальный клапан (30–50 %) и менее чем в 5 % случаев встречается дилатация корня аорты. Считают, что аневризма корня аорты, встречающаяся у больных с СШТ, наиболее вероятно, связана с дефектом мезенхимальной ткани, а не с атеросклеротическими изменениями. Синдром гипоплазии левых отделов сердца, который также является частым проявлением СШТ, несет высокий риск летальности. Считают, что данная аномалия преимущественно ассоциирована с кариотипом 45,X. При СШТ наблюдаются и такие сосудистые нарушения, как аберрантная правая подключичная артерия, недостаточность клапанов аорты, аортальный стеноз, недостаточность трехстворчатого клапана, пролапс митрального клапана [8, 19, 20, 23].
Генетические полиморфизмы, которые изменяют активность ферментов, участвующих в метаболизме фолиевой кислоты, в том числе метилентетрагидрофолатредуктазы (MTHFR), могут привести к гипометилированию ДНК. Ген MTHFR расположен на 1p36.3 и включает в себя два полиморфизма: замену цитозина на тимин в нуклеотиде 677 (C677T) и аденина на цитозин в нуклеотиде 1298 (A1298C), что приводит к снижению активности MTHFR и нарушению метаболизма фолиевой кислоты. Исследования полиморфизмов этого гена дали противоречивые результаты. Тем не менее показано, что применение фолиевой кислоты предотвращает развитие врожденных пороков сердца; уровень концентрации гомоцистеина в сыворотке крови коррелирует с развитием сердечно-сосудистых заболеваний, а дефицит половых гормонов у женщин является важным фактором, способствующим повышению содержания гомоцистеина [38].
В настоящее время существует дефицит осведомленности врачей и пациентов с СШТ о риске расслаивающей аневризмы аорты. К сожалению, пациенты часто игнорируют первые симптомы этого крайне неблагоприятного события, что значительно увеличивает риск летального исхода. В связи с этим считают, что у пациентов с СШТ необходимо проведение МРТ для своевременного оперативного вмешательства [39].
Fahrettin Oz и соавт. [29] при проведении допплеровского исследования внутрисердечного кровотока установили, что у больных с СШТ достоверно выше индекс массы миокарда левого желудочка (ИММЛЖ), толщина задней стенки ЛЖ, диаметр левого предсердия. Пик скорости ТМП в конце диастолы (пик А) был значительно выше, в то время как пик скорости трансмитрального потока в начале диастолы (пик Е), время замедления раннего трансмитрального кровотока и соотношение скоростей периодов раннего и позднего диастолического наполнения левого желудочка были значительно ниже у пациентов с СШТ.
Изменения сердечного ритма и метаболические нарушения в миокарде, подтвержденные результатами ЭКГ, были нами отмечены в 35,6 % случаев СШТ. Исследование электрической работы сердца выявило у больных с СШТ следующие нарушения: отклонение оси сердца вправо, нарушения зубца Т, блокада левой ножки пучка Гиса, укорочение интервала PR, удлинение интервала QT. Наиболее частым электрокардиографическим признаком, сопровождающим СШТ, являлся синдром удлинения интервала QT. Согласно данным Robert Dalla Pozza и соавт. [17], удлинение интервала QT наблюдается у 33 % больных СШТ в детском возрасте. Синдром удлинения интервала QT ассоциирован с высоким риском внезапной смерти. Учитывая тот факт, что у пациентов с СШТ наблюдается увеличение потенциала кардиомиоцитов дифференцироваться из плюрипотентных стволовых клеток, считают, что в основе электрофизиологических особенностей сердечной деятельности при моносомии X-хромосомы лежит первичный дефект развития [24]. По всей вероятности, синдром удлинения интервала QT у больных с СШТ обусловлен мутациями главных генов LQTS (Long QT syndrome) — KCNQ1, KCNH2 (c.3140G > Theterozygousp.Arg1047Leu, c.2738C > Theterozygousp.Ala913Val), KCNE1, KCNE2 (c.161T > Cheterozygousp.Met54Thr) и SCN5A (c.6010T > Cheterozygousp.Phe2004Leu, c.5872C > Theterozygousp.Arg1958X). Существует предположение о том, что SNP c.3140G > T гена KCNH2 (potassium channel, voltage gated eagrelated subfamily H, member 2) субъединицы aльфа-потенциалзависимого калиевого канала может быть связан с особой восприимчивостью к явлениям гипокалиемии и к действию некоторых лекарственных средств. SNP c.5872C > T гена SCN5A (sodium channel, voltage gated, type V alpha subunit) субъединицы бета-потенциалзависимого натриевого канала вызывает образование стоп-кодона. Мутация c.6010T > C гена SCN5A ассоциирована с внезапной смертью в младенческом возрасте. SNP c.161T > C гена KCNE2 (potassium channel, voltage gated subfamily E regulatory beta subunit 2) субъединицы бета-потенциалзависимого калиевого канала ассоциирована с желудочковой тахикардией, которая индуцирована физической нагрузкой [37].
Лекарственные средства, понижающие уровень холестерина, не влияют на продолжительность интервала QT у взрослых больных с СШТ. Антигипертензивными препаратами выбора у данных пациентов являются бета-блокаторы [5].
Christian Trolle и соавт. [37] предполагают, что в реализации синдрома удлиненного интервала QT у больных с СШТ определенную роль играет повышенный тонус симпатического отдела вегетативной нервной системы, который, вероятно, способствует повышению уровня возбудимости и проводимости проводящей системы сердца.
Следует отметить, что у 6 (17 %) детей отмечалась тахикардия и повышение артериального давления в покое без патологических ЭКГ и ЭхоКГ-изменений.
При проведении исследования липидного профиля у больных с СШТ нами было отмечено повышение коэффициента атерогенности в 9 (21,4 %) случаях, снижение липопротеидов высокой плотности (ЛПВП) — в 8 (19,1 %), повышение концентрации липопротеидов низкой плотности (ЛПНП) — в 6 (14,2 %), реже отмечалось повышение уровня холестерина и триглицеридов — в 5 (11,1 %) случаях.
Özgür Pirgon и соавт. [31] также продемонстрировали, что для детей с СШТ характерны высокие уровни липидов (общего холестерина и триглицеридов) в сыворотке крови. Гиперхолестеринемия диагностируется почти у 37 % взрослых с СШТ, т.е. в два раза чаще, чем в общей популяции. Уровень ЛПВП ниже 1,0 ммоль/л, который является бесспорным фактором риска развития атеросклероза, наблюдается у 25 % больных с СШТ и только у 5 % людей без хромосомной патологии [18].
Дислипидемия является определяющим фактором развития раннего атеросклероза у детей с СШТ [18].
Выводы
1. Дети с синдромом Шерешевского — Тернера в 80,9 % случаев имеют врожденные пороки развития и различные варианты нарушения функции сердечно-сосудистой системы. Наиболее часто встречаемыми врожденными пороками сердца при синдроме Шерешевского — Тернера являются коарктация аорты, двустворчатый аортальный клапан и дефект межпредсердной перегородки.
2. Синдром Шерешевского — Тернера в 35,6 % случаев сопровождается нарушениями электрической работы сердца, из которых наиболее прогностически неблагоприятным можно считать синдром удлиненного интервала QT.
3. Каждый пятый ребенок с синдромом Шерешевского — Тернера характеризуется изменением липидного профиля, который ассоциирован с риском развития раннего атеросклероза.
4. Рекомендуется систематическое, с периода новорожденности, проведение ЭКГ, ЭхоКГ, а при необходимости — МРТ сердца и крупных сосудов как для ранней диагностики врожденных пороков, нарушений электрической работы сердца, так и для своевременной медикаментозной коррекции или назначения оперативного лечения.
5. У детей с синдромом Шерешевского — Тернера следует регулярно контролировать липидный профиль с последующей модификацией диеты и физической нагрузки.
1. Берешева А.К. Мозаичная форма синдрома Шерешевского — Тернера с кольцевой хромосомой X у девочки 8 лет: применение методов молекулярно-цитогенетической диагностики / А.К. Берешева, И.Ю. Юров, А.Д. Колотый, И.М. Новикова, Ю.Б. Юров, С.Г. Ворсанова // Российский вестник перинатологии и педиатрии. — 2011. — Т. 56, № 5. — С. 30-37.
2. Вяткина С.В. Современные представления о синдроме Шерешевского — Тернера / С.В. Вяткина, Т.В. Кузнецова // Журнал акушерства и женских болезней. — 2007. — Т. LVI, № 1. — С. 56-63.
3. Драгун С.А. Оценка состояния минеральной плотности костной ткани, костного метаболизма, углеводного обмена, органов репродуктивной системы и иммунного статуса у больных с синдромом Шерешевского — Тернера в разные возрастные периоды: Автореф. дис… канд. мед. наук. — М., 2006. — 23 с.
4. Панкратова М.С. Синдром Шерешевского — Тернера: особенности мониторинга в разные возрастные периоды / М.С. Панкратова, В.А. Петеркова // Международный эндокринологический журнал. — 2010. — 6 (30). — С. 83-86.
5. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology / American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) / European Heart Rhythm Association; Heart Rhythm Society, Zipes D.P., Camm A.J., Borggrefe M., Buxton A.E., Chaitman B., Fromer M., Gregoratos G., Klein G., Moss A.J., Myerburg R.J., Priori S.G., Quinones M.A., Roden D.M., Silka M.J., Tracy C., Smith S.C. Jr., Jacobs A.K., Adams C.D., Antman E.M., Anderson J.L., Hunt S.A., Halperin J.L., Nishimura R., Ornato J.P., Page R.L., Riegel B., Priori S.G., Blanc J.J., Budaj A., Camm A.J., Dean V., Deckers J.W., Despres C., Dickstein K., Lekakis J., McGregor K., Metra M., Morais J., Osterspey A., Tamargo J.L., Zamorano J.L.; American College of Cardiology; American Heart Association Task Force; European Society of Cardiology Committee for Practice Guidelines // J. Am. Coll. Cardiol. 2006 Sep 5; 48 (5): e247-346.
6. Al Alwan I. Turner Syndrome Genotype and phenotype and their effect on presenting features and timing of Diagnosis / I. Al Alwan, M.K., Amir 1st3, G.N., A.O., L.B., M.A.D., M.B. // Int. J. Health Sci (Qassim). 2014 Apr; 8 (2): 195-202. PMCID: PMC4166992.
7. Baena N., De Vigan C., Cariati E. et al. Turner syndrome: Evaluation of prenatal diagnosis in 19 European registries // Am. J. Med. Genet. 2004; 129A: 16-20.
8. Baguet J.P. Structural and functional abnormalities of large arteries in the Turner syndrome/ J.P. Baguet, S. Douchin, H. Pierre еt al. // Heart. 2005 Nov; 91 (11): 1442-6. doi:10.1136/hrt.2004.048371.
9. Baş F. The exon 3-deleted/full-length growth hormone receptor polymorphism and response to growth hormone therapy in growth hormone deficiency and Turner syndrome: a multicenter study / F. Baş, F. Darendeliler, Z. Aycan еt al. // Horm. Res. Paediatr. 2012; 77 (2): 85-93. doi: 10.1159/000335172.
10. Bianco B. Analysis of vitamin D receptor gene (VDR) polymorphisms in Turner syndrome patients / Bianco B., Verreschi I.T., Oliveira K.C. еt al. // Gynecol. Endocrinol. 2012 Apr; 28 (4): 326-9. doi: 10.3109/09513590.2011.631630.
11. Bianco B. PTPN22 polymorphism is related to autoimmune disease risk in patients with Turner syndrome / B. Bianco, I.T. Verreschi, K.C. Oliveira еt al. // Scand. J. Immunol. 2010 Sep; 72 (3): 256-9. doi: 10.1111/j.1365-3083.2010.02438.x.
12. Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy // Horm. Res. Paediatr. 2011 Feb; 75 (2): 81-9. doi: 10.1159/000324105.
13. Bondy C.A. Heart disease in Turner syndrome // Minerva Endocrinol. 2007 Dec; 32 (4): 245-61. PMID: 18091662.
14. Bondy C.A. Turner’s syndrome and X chromosome-based differences in disease susceptibility // Gend. Med. 2006 Mar; 3 (1): 18-30.
15. Braz A.F. Genetic predictors of long-term response to growth hormone (GH) therapy in children with GH deficiency and Turner syndrome: the influence of a SOCS2 polymorphism / A.F. Braz, E.F. Costalonga, E.B. Trarbach еt al. // J. Clin. Endocrinol. Metab. 2014 Sep; 99 (9): E1808-13. doi: 10.1210/jc.2014-1744.
16. Braz A.F. The interactive effect of GHR-exon 3 and -202 A/C IGFBP3 polymorphisms on rhGH responsiveness and treatment outcomes in patients with Turner syndrome / A.F. Braz, E.F. Costalonga, L.R. Montenegro еt al. // J. Clin. Endocrinol. Metab. 2012 Apr; 97 (4): E671-7. doi: 10.1210/jc.2011-2521.
17. Dalla Pozza R. QTc interval prolongation in children with Ulrich-Turner syndrome / R. Dalla Pozza, S. Bechtold, S. Kääb еt al. // Eur. J. Pediatr. 2006 Dec; 165 (12): 831-7. Epub 2006 Jul 12.
18. Elsheikh M. Turner’s syndrome in adulthood / M. Elsheikh, D.B. Dunger, G.S. Conway, J.A. Wass // Endocr. Rev. 2002 Feb; 23 (1): 120-40. doi: http://dx.doi.org/10.1210/edrv.23.1.0457.
19. Frías J.L., Davenport M.L. Health supervision for children with Turner syndrome // Pediatrics. 2003 Mar; 111 (3): 692-702. doi: 10.1542/peds.111.3.692.
20. Gravholt C.H. Clinical and epidemiological description of aortic dissection in Turner's syndrome / C.H. Gravholt, K. Landin-Wilhelmsen, K. Stochholm еt al. // Cardiol Young. 2006 Oct; 16 (5): 430-6. doi: http://dx.doi.org/10.1017/S1047951106000928.
21. Hook E.B., Warburton D. Turner syndrome revisited: review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss // Hum. Genet. 2014 Apr; 133 (4): 417-24. doi: 10.1007/s00439-014-1420-x.
22. Ko J.M. The common exon 3 polymorphism of the growth hormone receptor gene and the effect of growth hormone therapy on growth in Korean patients with Turner syndrome / J.M. Ko, J.M. Kim, C.K. Cheon еt al. // Clin. Endocrinol. (Oxf). 2010 Feb; 72 (2): 196-202. doi: 10.1111/j.1365-2265.2009.03681.x.
23. Kriksciuniene R., Ostrauskas R., Zilaitiene B. Aortopathies in Turner syndrome — new strategies for evaluation and treatment // Endokrynol Pol. 2015; 66 (1): 58-65. doi: 10.5603/EP.2015.0010.
24. Li W. Modeling abnormal early development with induced pluripotent stem cells from aneuploid syndromes / W. Li, X. Wang, W. Fan еt al. // Hum. Mol. Genet. 2012 Jan 1; 21 (1): 32-45. doi: 10.1093/hmg/ddr435.
25. Lucaccioni L. Turner syndrome-issues to consider for transition to adulthood / L. Lucaccioni, S.C. Wong, A. Smyth еt al. // Br. Med. Bull. 2015 Mar; 113 (1): 45-58. doi: 10.1093/bmb/ldu038.
26. Milbrandt T., Thomas E. Turner syndrome // Pediatr. Rev. 2013 Sep; 34 (9): 420-1. doi: 10.1542/pir.34-9-420.
27. Oliveira K.C. C677T and A1298C polymorphisms of MTHFR gene and their relation to homocysteine levels in Turner syndrome / K.C. Oliveira, I.T. Verreschi, E.K. Sugawara еt al. // Genet. Test Mol. Biomarkers. 2012 May; 16 (5): 396-400. doi: 10.1089/gtmb.2011.0222.
28. Oliveira K.C. Prevalence of the polymorphism MTHFR A1298C and not MTHFR C677T is related to chromosomal aneuploidy in Brazilian Turner Syndrome patients / K.C. Oliveira, B. Bianco, I.T. Verreschi еt al. // Arq. Bras. Endocrinol. Metabol. 2008 Nov; 52 (8): 1374-81. http://dx.doi.org/10.1590/S0004-27302008000800028/
29. Oz F. Doppler-derived strain imaging detects left ventricular systolic dysfunction in children with turner syndrome / F. Oz, A.Y. Cizgici, A. Ucar еt al. // Echocardiography. 2014 Sep; 31 (8): 1017-22. doi: 10.1111/echo.12500.
30. Peralta Lуpez M. Association of vitamin D receptor gene Cdx2 polymorphism with bone markers in Turner syndrome patients / Peralta Lуpez M., Centeno V., Miras M. еt al. // J. Pediatr. Endocrinol. Metab. 2012; 25 (7-8): 669-71. doi: 10.1515/jpem-2012-0098.
31. Özgür Pirgon. Atherogenic lipid profile and systolic blood pressure are associated with carotid artery intima-media thickness in children with Turner syndrome / Özgür Pirgon, M.E. Atabek, B. Oran, R. Güзlü // J. Clin. Res. Pediatr. Endocrinol. 2008; 1 (2): 62-71. doi: 10.4008/jcrpe.v1i2.9.
32. Poprawski K. Cardiovascular abnormalities in patients with Turner syndrome according to karyotype: own experience and literature review / K. Poprawski, M. Michalski, M. Јawniczak, K. Јacka // Pol. Arch. Med. Wewn. 2009 Jul-Aug; 119 (7-8): 453-60.
33. Prakash S. Single-nucleotide polymorphism array genotyping is equivalent to metaphase cytogenetics for diagnosis of Turner syndrome / S. Prakash, D. Guo, C.L. Maslen еt al.; GenTAC Investigators // Genet. Med. 2014 Jan; 16 (1): 53-9. doi: 10.1038/gim.2013.77.
34. Sowiсska-Przepiera E. Association between ER polymorphisms and bone mineral density in patients with Turner syndrome subjected to estroprogestagen treatment — a pilot study / E. Sowiсska-Przepiera, E. Andrysiak-Mamos, K. Cheіstowski еt al. // J. Bone Miner. Metab. 2011 Jul; 29 (4): 484-92. doi: 10.1007/s00774-010-0247-3.
35. Struwe E. No evidence for angiotensin type 2 receptor gene polymorphism in intron 1 in patients with coarctation of the aorta and Ullrich-Turner syndrome / E. Struwe, K. Krammer, J. Dötsch еt al. // Pediatr. Cardiol. 2006 Sep-Oct; 27 (5): 636-9.
36. Sybert V.P., McCauley E. Turner’s syndrome // N. Engl. J. Med. 2004 Sep 16; 351 (12): 1227-38. doi: 10.1056/NEJMra030360.
37. Trolle C. Long QT interval in Turner syndrome — a high prevalence of LQTS gene mutations / C. Trolle, K.H. Mortensen, L.N. Pedersen at al. // PLoS One. 2013 Jul 25; 8 (7): e69614. doi: 10.1371/journal.pone.0069614.
38. Trove-Marqui A.B. Sindrome de Turner e polimorfismo genetico: umarevisio sistematica // Rev. Paul. Pediatr. 2015 Feb 18. pii: S0103-0582 (15)00015-5. doi: 10.1016/j.rpped.2014.11.014.
39. Turtle E.J. Aortic dissection in children and adolescents with Turner syndrome: risk factors and management recommendations / E.J. Turtle, A.A. Sule, D.J. Webb, L.E. Bath // Arch. Dis. Child. 2015 Jan 8. pii: archdischild-2014-307080. doi: 10.1136/archdischild-2014-307080.
40. Yeëilkaya E. Turner Syndrome and Associated Problems in Turkish Children: A Multicenter Study / E. Yeëilkaya, A. Bereket, F. Darendeliler еt al. // J. Clin. Res. Pediatr. Endocrinol. 2015 Mar 5; 7 (1): 27-36. doi: 10.4274/jcrpe.1771.
41. Zhong Q., Layman L.C. Genetic considerations in the patient with Turner syndrome-45,X with or without mosaicism // Fertil. Steril. 2012 Oct; 98 (4): 775-9. doi: 10.1016/j.fertnstert.2012.08.021.