Статья опубликована на с. 128-132
В структуре заболеваний гепатобилиарной системы у детей первого полугодия жизни ведущее место занимает билиарная атрезия (45 %), далее по частоте встречаемости располагаются синдром Алажиля (17 %), болезнь Байлера (13 %), неонатальный гепатит (5 %), дефицит α1-антитрипсина (4 %), билиарный цирроз (4 %), галактоземия (2 %) и другие заболевания (10 %) [1]. Синдром холестаза является одним из ранних проявлений широкого спектра заболеваний гепатобилиарной системы у новорожденных и детей первых месяцев жизни.
Артериопеченочная дисплазия (синдром Алажиля) включает сочетание не менее трех из пяти основных признаков: хронический холестаз, сердечно-сосудистые дефекты, аномалии позвоночника, дефекты глаз, характерные черепно-лицевые признаки. Встречается с частотой 1 : 70 000 новорожденных. Синдром впервые описан французским педиатром Даниелем Алажилем в 1975 году как заболевание с типичной комбинацией пяти признаков:
— холестаза;
— лицевого дисморфизма;
— незаращения тел преимущественно грудных позвонков с характерной картиной «бабочки» на рентгенограмме;
— стеноза легочной артерии и/или ее ветвей и других пороков сердца;
— офтальмологических аномалий — заднего эмбриотоксона (врожденный дефект роговицы), пигментной ретинопатии.
Синдром Алажиля наследуется по аутосомно-доминантному типу. Генный дефект связан с частичной делецией короткого плеча 20-й хромосомы [20р11-12], где локализуется Jagged1 (JAG1) ген, что доказано молекулярно-генетическими и цитогенетическими методами исследования.
Болезнь характеризуется недостаточным количеством или малым диаметром внутрипеченочных желчных протоков, что затрудняет отток желчи и способствует накоплению ее компонентов в клетках печени. Повышенное содержание желчных компонентов в плазме крови способствует появлению мучительного кожного зуда. Недостаточное поступление желчи в кишечник приводит к нарушению процессов пищеварения. Мальабсорбция ведет к задержке развития и медленному росту. У таких больных часто возникают переломы костей, проблемы со зрением, свертываемостью крови, памятью и обучением. Характерна задержка полового развития. Явления холестаза развиваются в первые 3 месяца жизни и характеризуются желтухой с зеленоватым оттенком, увеличением размеров печени, непостоянной ахолией стула, темным цветом мочи. Печень увеличивается больше за счет левой доли, которая при пальпации гладкая, безболезненная, с закругленным краем, консистенция ее нормальная или умеренно плотная. Часто увеличивается селезенка. При синдроме Алажиля под кожей могут образовываться жировые скопления, называемые ксантомами (на тыльной поверхности суставов пальцев, на ладонной поверхности кистей рук, на задней поверхности шеи, в паховой области, вокруг ануса). Они вызваны аномально высоким уровнем холестерина в крови и свидетельствуют о тяжести процесса и длительности холестаза. Выявляются характерные фенотипические особенности: высокий, выступающий лоб, глубоко посаженные глаза, прямой нос, оттопыренные ушные раковины, выступающий подбородок. У больных с синдромом Алажиля выявляются изменения со стороны почек — гипоплазия, кисты, дистопия почек, удвоение мочеточника, стеноз почечной артерии, мочекаменная болезнь [2–4].
Специфическая терапия не разработана. Лечение направлено на поддержание функций пораженных органов и уменьшение симптомов заболевания. Для улучшения функции печени применяют гепатопротекторные препараты; для уменьшения желтухи и зуда — препараты урсодезоксихолевой кислоты. При тяжелом течении проводят трансплантацию печени [3].
Если атрезия внутрипеченочных желчных ходов выражена незначительно (отсутствует лишь небольшое количество желчных ходов), прогноз благоприятный и осложнения обычно не возникают. В большинстве случаев атрезия значительная, что является неблагоприятным для прогноза; в течение первых лет жизни развивается цирроз печени — больные погибают от печеночной недостаточности. Трансплантация печени позволяет продлить больным жизнь, но в случае сочетания с тяжелым пороком сердца ее проведение невозможно [5–6].
Приводим собственное наблюдение.
Мальчик Б., 11 месяцев, поступил в гастроэнтерологическое отделение с жалобами на интенсивную желтушность кожи и слизистых с зеленоватым оттенком, вялость, рвоту, выпадение прямой кишки.
Из анамнеза жизни и болезни известно, что ребенок родился от 1-й беременности, протекавшей с преэклампсией. Роды первые, самопроизвольные, в срок. Вес при рождении 2700 г. Период новорожденности протекал гладко, но мать отмечала желтушность кожных покровов с рождения, плохую прибавку в весе. В возрасте 1,5 месяца на фоне сохраняющейся желтушности кожи появился разжиженный стул. Ребенок госпитализирован в центральную районную больницу, где при осмотре выявлены повышенная кровоточивость, экхимозы. Мальчик был направлен в гематологическое отделение городской детской клинической больницы, где находился в течение 2 недель; диагностирован криптогенный гепатит неустановленной этиологии с выраженной степенью активности, вегето-висцеральная дисфункция на фоне перинатальной энцефалопатии. Для обследования переведен в областную детскую инфекционную клиническую больницу (ОДИКБ). В стационаре ребенок находился в течение месяца, был установлен диагноз: врожденная цитомегаловирусная инфекция, гепатит, тяжелая форма. Проводилась комплексная терапия, включая противовирусную. Однако мать самовольно забрала ребенка домой, оставив без лечения. Через месяц с жалобами на повышение температуры тела до 38,5 °С, нарастание желтухи повторно госпитализируется в ОДИКБ. Исследование на маркеры вирусной инфекции методом ПЦР — HBV, HCV, ЦМВ, ВЭБ — отрицательное. При проведении иммуноферментного анализа (ИФА) антитела классов IgG, IgМ к ВПГ 6-го типа, токсоплазмозу, IgG к ЦМВ, ВЭБ-инфекции — отрицательные. Для исключения атрезии внутрипеченочных желчных протоков ребенок переведен в хирургический стационар, где данная патология исключена. При выписке общий билирубин 112 мкмоль/л (прямой 63,4; свободный 48,6), АЛТ до 11 норм, АСТ до 10 норм. Рекомендовано: продолжить прием урсофалька, атоксила, вскармливание низколактозными смесями, наблюдение педиатра. Спустя три месяца повысилась вялость, сонливость, желтушность кожных покровов приобрела зеленоватый оттенок, появилась полифекалия и осветление кала. Для проведения диагностической лапароскопии и биопсии печени направлен в хирургическое отделение многопрофильного областного стационара. Диагностирован хронический гепатит с формированием мелкоузлового цирроза печени и рекомендовано обследование и лечение в гастроэнтерологическом отделении многопрофильного областного стационара.
При поступлении состояние очень тяжелое за счет терминальной стадии печеночной недостаточности. Гипотрофия 2-й степени (вес 7 кг). В сознании, на осмотр реагирует негативно, двигательная активность снижена, общая вялость, гипотония мышц, задержка психомоторного развития (самостоятельно не сидит). Выражена желтушность кожи, слизистых и склер с зеленоватым оттенком. Пастозность лица, стоп, голеней. Лицо треугольной формы с широким выпуклым лбом, глубоко посаженными глазами, уплощенным, «птичьим» носом и острым подбородком, искривление V пальцев рук. В легких дыхание пуэрильное, хрипов нет. Границы сердца не изменены. Тоны сердца ритмичные, громкие, систолический шум на верхушке. Живот увеличен в размерах, асцит. Печень пальпировалась до 3 см по среднеключичной линии, плотной консистенции, селезенка до 2,5 см. Стул окрашенный, светло-желтый, полифекалия, выпадение прямой кишки. Моча темная.
Данные дополнительных исследований
Клинический анализ крови: гемоглобин — 75 г/л, эр. — 2,5 • 1012/л, ЦП — 0,8, тромбоциты — 70 • 109/л, лейкоциты — 22 · 109/л, палочкоядерные нейтрофилы — 1 %, сегментоядерные нейтрофилы — 55 %, лимф. — 36 %, мон. — 7 %, СОЭ — 5 мм/час.
Гематокрит — 18 %.
Клинический анализ мочи: кол-во — 100, цвет желтый, прозр. умерен., уд. вес 1020, белок, глюкоза — не обнаружены, реакция кислая, эпителий переходный 1–2 в п/зр., лейкоциты — 1–2 в п/зр.
Гельминты и простейшие в кале не обнаружены.
Копрограмма — стеаторея.
Биохимический анализ крови: цитолиз (АЛТ 10 норм, АСТ 12 норм, гипербилирубинемия 403 мкмоль/л, связанный билирубин 294 (норма 2,1–6,4 мкмоль/л), свободный билирубин 108 мкмоль/л (норма 6,5–19,1 мкмоль/л), повышение уровня щелочной фосфатазы в 2 нормы).
Выявление синдрома холестаза диктует необходимость дифференциальной диагностики между вторичными нарушениями и заболеваниями гепатобилиарной системы (врожденная тирозинемия, болезнь Байлера, неполная атрезия желчных протоков, вторичный билиарный цирроз на фоне перенесенной генерализованной ЦМВ-инфекции, гликогенозы, пероксисомные заболевания, другие аминоацидопатии, болезнь Коновалова — Вильсона, муковисцидоз, дефицит альфа-1-антитрипсина). В связи с этим проведены нижеперечисленные исследования:
Протеинограмма: общий белок — 51 г/л, альбумин — 44 %.
Коагулограмма: протромбиновый индекс — 70,8 % (норма 93–107 %), время свертывания — 13 мин (норма 5–10 минут).
Альфа-1-антитрипсин — в норме.
Альфа-фетопротеин — более 100 000 нг/мл (норма < 11,3), в динамике — 3600 нг/мл. Дефицит желчных протоков может компенсироваться их пролиферацией, что ассоциировано с высокой экспрессией альфа-фетопротеина.
Гамма-глутаминтрансфераза (ГГТ) — 0,9 (норма 0,9–6,36 ммоль/час Ч л).
Исследование на маркеры вирусной инфекции методом ПЦР — IgM к ЦМВ — отр., IgG к ЦМВ — 2,05 (норма < 0,5). Данные изменения расценены как персистирующая инфекция, вызванная вирусами ЦМВ.
Глюкоза крови — 2,8 ммоль/л.
Скрининг-тесты мочи — в норме.
Пилокарпиновая проба — в норме.
Медь мочи — в норме.
Церулоплазмин — в норме.
УЗИ органов брюшной полости и почек: печень увеличена на 5 см, эхогенность паренхимы повышена, холестаз, воротная вена дилатирована до 8 мм, извита. Желчный пузырь: удлинен, гипотоничен, стенка отечна до 3–4 мм, полость свободна. Селезенка увеличена на 4,5 см, край утолщен, токсико-пролиферативная реакция паренхимы, вены умеренно дилатированы. Почки: уростаза нет, паренхима не изменена.
УЗИ головного мозга: отечность оболочек, рыхлость эпендимы, межполушарная щель 2 мм.
Компьютерная томография пояснично-крестцового отдела позвоночника: КТ-признаки аномалии развития пояснично-крестцового перехода (люмбализация L6, spina bifida L6), агенезия копчика.
ЭКГ: синусовая брадикардия, выраженные нарушения реполяризации миокарда.
ДпЭхоКГ: лево-правый шунт диаметром 4,8–5,5 мм в центральной части межпредсердной перегородки (МПП). Дилатация правых камер. Трикуспидальная регургитация 1-й степени. Заключение: врожденный порок сердца; вторичный дефект МПП.
Рентгенография органов грудной клетки: легочный рисунок не изменен. Корни — за тенью средостения, расширенного за счет тимомегалии 1-й ст., синусы свободны. Сердце расширено в поперечнике, КТИ — 58 %.
Патологогистологическое исследование биоптата печени: два участка ткани коричневато-зеленоватого цвета с фрагментом капсулы по 0,6 × 0,3 × 0,5 см, плотной консистенции. Заключение: хронический гепатит с формированием мелкоузлового цирроза.
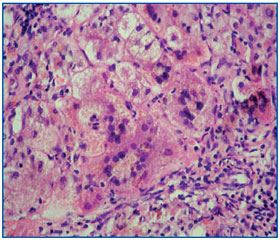
Патоморфологическое исследование биоптата печени (окраска гематоксилин-эозином, увеличение × 200): биоптат печени с нарушением структуры ткани за счет образования множества ложных долек, с гигантоклеточным метаморфозом гепатоцитов. Дольки разделены широкими соединительнотканными септами с неравномерной мононуклеарно-лейкоцитарной инфильтрацией (рис. 1).
Патоморфологическое исследование биоптата печени (окраска гематоксилин-эозином, увеличение × 400): гигантоклеточная трансформация гепатоцитов. Признаки баллонирующей дистрофии гепатоцитов (коликвационный некроз) (рис. 2).
Консультирован лор-врачом — патологии не выявлено; окулистом — патологии не выявлено; неврологом — задержка темпов психомоторного развития вследствие перинатального гипоксически-ишемического поражения ЦНС; кардиолог, кардиохирург — врожденный порок сердца, вторичный дефект межпредсердной перегородки, НКО. Данных в пользу легочной гипертензии нет.
Заключение генетика: у ребенка выявлены сложные нарушения в обмене веществ — нарушение обмена длинноцепочечных жирных кислот, серосодержащих аминокислот.
Сочетание внутриутробной гипотрофии, гипоплазии внутрипеченочных желчных протоков, ВПС (ДМПП), биохимических маркеров холестаза, изменений со стороны опорно-двигательного аппарата (лицевые дисморфии, признаки аномалии развития пояснично-крестцового перехода (люмбализация L6, spina bifida L6), агенезия копчика) и отставание в физическом развитии стали основанием для диагноза: cиндром Алажиля. Вторичный билиарный цирроз печени, мелкоузловой. Хроническая печеночная недостаточность с нарушением белково-синтетической функции печени, терминальная стадия. Нарушение обмена серосодержащих аминокислот, длинноцепочечных жирных кислот.
На фоне терапии гепатита и синдрома холестаза состояние ребенка оставалось тяжелым: сохранялась выраженная желтушность кожи, слизистых, склер, периодически неустойчивый стул, вялость, плохая прибавка в весе, гипербилирубинемия. С учетом характера заболевания, развития цирроза печени ребенок был направлен на консультацию в Национальный институт хирургии и трансплантологии АМН Украины им. проф. А.А. Шалимова.
Заключение хирурга-трансплантолога: терминальная стадия заболевания печени, внутрипеченочный холестаз, синдром Алажиля. Рекомендована пересадка печени.
Трансплантация печени ребенку не проведена в связи с материальными трудностями семьи. Ухудшение состояния ребенка на фоне прогрессирования заболевания наступило через 2 месяца. Госпитализирован в клинику.
Состояние ребенка при поступлении тяжелое, что обусловлено хронической печеночной недостаточностью в терминальной стадии. При осмотре в сознании, резко вялый, адинамичный, постанывает. Не лихорадит. Катаральных явлений нет. Одышка выражена минимально, смешанного характера, ЧД 28–30 в 1 минуту, ЧСС от 104 до 120 ударов в 1 минуту. Кожа, склеры, слизистые желтушные с зеленоватым оттенком. Пастозность век, стоп, передней брюшной стенки. В легких дыхание жесткое, хрипов нет. Тоны сердца приглушенные, ритмичные, систолический шум над областью сердца. Живот увеличен в размерах, вздут. Печень до 13 см ниже края реберной дуги, селезенка до 4,0 см. Стул светло-желтый, кашицеобразный. Периодически выпадает прямая кишка. Моча желтая. При обследовании общий билирубин 1005,8 мкмоль/л (прямой — 578,0; свободный — 427,8), значительное повышение уровня трансаминаз: АЛТ — 1,88, АСТ — 1,55 (N — 0,06–0,14). Проводилась терапия, включающая урсохол, глутаргин, сорбенты, диуретики, витамины, ферменты, эубиотики, антибактериальные препараты, инфузию глюкозо-солевых растворов с целью дезинтоксикации, коррекцию гипопротеинемии (10% альбумин), дизэлектролитных нарушений; в дальнейшем с целью повышения коагуляционного потенциала крови проводились инфузии свежезамороженной плазмы и коррекция анемии трансфузией тромбоцитарной массы. Несмотря на проведение комплексной терапии, на 3-и сутки состояние больного прогрессивно ухудшается на фоне терминальной стадии хронической печеночной недостаточности. Ребенок становится резко адинамичным, сонливым, изредка открывает глаза, прогрессирует печеночная энцефалопатия. Обращает на себя внимание печеночный запах. Кожные покровы, видимые слизистые, склеры резко-желтушно-зеленоватого цвета, сухость кожи, зуд. Отмечаются подъемы температуры тела до фебрильных цифр. Аппетит снижен. Отмечается тошнота, повторная рвота. Нарастает отечный синдром. Присоединяется ДВС-синдром, который в последние сутки жизни прогрессивно нарастает. Выражен парез кишечника, повторяются выпадения прямой кишки. Увеличивается гипербилирубинемия, уровень транаминаз остается высоким, несмотря на повторные инфузии 10% альбумина, сохраняется гипопротеинемия, гипоксемия трудно поддается коррекции, прогрессируют тромбоцитопения, изменения свертывающей системы крови, воспалительные изменения в крови, регистрируется декомпенсированный метаболический ацидоз. В динамике наблюдения на фоне терминальной стадии печеночной недостаточности прогредиентно нарастают проявления полиорганных нарушений — церебральная, сердечно-сосудистая, дыхательная недостаточность, ДВС-синдром. На 3-и сутки пребывания в стационаре зарегистрирована биологическая смерть.
Патологоанатомический диагноз: МВПР: 1. Синдром Алажиля (внутрипеченочный холестаз, ВПС — ІІ АВД, аномалия развития позвоночника (люмбализация L6, агенезия копчика)). 2. Нарушение обмена серосодержащих аминокислот, длинноцепочечных жирных кислот.
Осложнения: вторичный билиарный цирроз печени, мелкоузловой. Хроническая печеночная недостаточность с нарушением белково-синтетической функции печени, терминальная стадия. Вторичная митохондриальная дисфункция.
Сопутствующий: вторичная КМП, НК ІІ АБ. Гипотрофия ІІ ст., пре- и постнатальная, смешанного генеза. Выпадение прямой кишки, анемия тяжелой степени.
Таким образом, диагностика синдрома Алажиля основана на выявлении характерных особенностей фенотипа и 2 или более типичных аномалий и/или пороков развития других органов и/или систем. Прогностическое значение при динамическом обследовании детей с синдромом Алажиля имеет уровень билирубина, нормализация которого во втором полугодии жизни свидетельствует о высокой вероятности отсутствия показаний к проведению трансплантации печени в течение длительного времени, возможно в течение всей жизни. Системность поражения при синдроме Алажиля осложняет течение болезни и затрудняет лечение. Выбор хирургической тактики зависит от множественных сочетанных пороков развития. Как один из вероятных этиологических факторов, приводящих к малому диаметру внутрипеченочных желчных протоков во внутриутробном периоде, можно рассматривать цитомегаловирус. Нарушение обмена серосодержащих аминокислот, длинноцепочечных жирных кислот не входит в комплекс изменений органов и систем при синдроме Алажиля и ухудшает прогноз заболевания ребенка.

/131-1.jpg)
/131-2.jpg)