Статтю опубліковано на с. 70-78
Ендотелій — орган внутрішньої секреції, що регулює тонус судин, захищає їх від негативної дії циркулюючих клітин і субстанцій, контролює транспортування розчинених речовин у клітини судинної стінки, здійснює контроль імунних, запальних і репаративних процесів, підтримує баланс місцевих процесів гемостазу [1, 2]. Зазначені функції ендотелій реалізує шляхом синтезу та виділення біологічно активних сполук: вазодилатуючих (оксид азоту (NO), простациклін, ендотеліальний фактор гіперполяризації) та вазоконстрикторних/протромбогенних (ендотелін-1, тромбоксан А2, простагландин Н2, ангіотензинперетворювальний фермент (АПФ) та утворюваний за його допомогою ангіотензин ІІ (А ІІ)).
Оксид азоту, найпотужніший серед зазначених вазодилататорів, є молекулою малого розміру, що являє собою високореактивний радикал завдяки наявності на зовнішній орбіті непарного електрона. Висока ліпофільність NO зумовлює його вільне проникнення через клітинні мембрани. NO синтезується з амiнокислоти L-аргiнiну трьома основними iзоформами NO-синтази, що названі відповідно до тих типів клітин, де вони були вперше знайдені: nNOS (нейрональна, NOS-1), mNOS (макрофагальна, або індуцибельна, NOS-2) та еNOS (ендотеліальна, NOS-3) [3]. Ці ферменти гомологічні лише на 50–60 % за своїм амінокислотним складом і кодуються різними генами. Ендотеліальна та нейрональна ізоформи NOS належать до конститутивного різновиду ферменту, індуцибельна NO-синтаза з’являється в клітинах після індукції її бактеріальними ендотоксинами, ліпополісахаридами, цитокінами, такими як інтерлейкін-1, інтерлейкін-2, фактор некрозу пухлини (TNF) та ін. [4]. Конститутивні форми активуються таким чином: Ca2+ під впливом певних стимулів входить у клітину, у цитозолі зв’язується в єдиний комплекс із кальмодуліном. Комплекс «кальцій — кальмодулін» виступає в ролі кофактора, активуючи NOS; у свою чергу, активована NOS синтезує обмежену кількість NO. До стимулів, що збільшують проникність Ca2+ в ендотеліоцити, належать: 1) пульсуючий кровообіг, що створює так звану напругу зсуву; 2) катехоламіни, що циркулюють із током крові (адреналін, норадреналін), ацетилхолін, вазопресин, що стимулюють α2-, М- та Vp-рецептори ендотеліоцитів відповідно; 3) речовини, що синтезуються судинною стінкою, — брадикінін через В2-рецептори та гістамін з Н-рецепторами; 4) продукти активації тромбоцитів — аденозин (Рy-рецептори) та серотонін (5НТ10-рецептори); 5) тромбін, що з’являється після запуску коагуляційного каскаду, стимулює Т-рецептори ендотеліоцитів [5].
Оксид азоту з ендотелiальної клiтини проникає в міоцити судинної стiнки, активує розчинну гуанiлатциклазу, що приводить до підвищення рівня циклічного гуанозинмонофосфату (цГМФ), активацiї цГМФ-залежних протеїнкiназ, зниження концентрації іонів кальцію та розслаблення судин [6, 7]. Отже, NO бере активну участь у регуляції судинного тонусу, регулює периферичний опір, артеріальний тиск і розподіл кровообігу в судинній мережі [8]. Вивільнення NO з ендотеліоцитів безпосередньо в просвіт судини перешкоджає адгезії тромбоцитів і лейкоцитів до ендотелію. Окрім зазначених функцій, NO здатний підтримувати гомеостаз судинної стінки шляхом синтезу ендотеліального фактора росту [9], стимуляції ангіогенезу, гальмування проліферації та міграції гладком’язових клітин [10].
У разі зниження біодоступності NO виникає ендотеліальна дисфункція (ЕД), що характеризується патологічними змінами вищезазначених властивостей ендотелію. У свою чергу, ЕД призводить до порушення роботи органів і систем. Так, при ЕД добре вивченими є зміни не лише серцево-судинної системи, а й нервової [11], нирок [12], легень [13].
Треба також зазначити, що уніфікованого загальноприйнятого способу дослідження порушення функції ендотелію немає. Останнім часом вивчаються методи оцінки ЕД за допомогою маркерів активації ендотеліоцитів (Е-селектин), їх пошкодження (фактор Віл-лебранда, клітини-попередники ендотеліоцитів, мікрочастки ендотеліоцитів тощо). Проте найбільш поширеними та випробуваними часом залишаються методи, що оцінюють ступінь дилатації судини шляхом внутрішньовенного введення ацетилхоліну (АХ) чи під впливом механічного чинника — проби з гіперемією. Остання, враховуючи її безпечність і доступність, розглядається як неінвазивний золотий стандарт оцінювання вазодилатуючої функції ендотелію [14].
З метою з’ясування ступеня значущості досліджуваного чинника в конкретній нозології традиційно вивчають його вплив на її клінічний прогноз. На сьогодні в галузі кардіології накопичено значну кількість досліджень, у яких ЕД вивчалася як такий предиктор. Зокрема, у хворих на гіпертонічну хворобу (ГХ) спостерігається зниження ендотелійзалежної вазодилатації (ЕЗВД) периферичних артерій [15, 16], парадоксальна вазоконстрикція коронарних артерій (КА) при пробі з АХ [17]. У 2264 жінок у менопаузі з підвищеним артеріальним тиском ЕД, що не коригувалася після 6 міс. антигіпертензивного лікування, асоціювалася з підвищенням частоти серцево-судинних подій (раптова серцева смерть, інфаркт міокарда (ІМ) протягом 45 міс. спостереження) [18]. У Northern Manhattan Study серед 819 осіб із метаболічним синдромом вивчали прогностичне значення вазодилатуючої функції ендотелію в пробі з реактивною гіперемією (РГ). Усі обстежувані мали високий ризик серцево-судинних подій (HR –1,5), тоді як серед осіб зі зниженою ЕЗВД він становив 2,6, що дозволило дослідникам зробити висновок про незалежну прогностичну цінність ЕД серед безсимптомних пацієнтів [19].
Відповідно до результатів 5-річного дослідження MESA, проведеного серед 3000 осіб літнього віку без задокументованої серцево-судинної патології, ЕЗВД є предиктором серцево-судинних подій (ІМ, інсульт, серцево-судинна смерть (ССС), стенокардія, виконання коронарної реваскуляризації). Після проведення мультиваріантного аналізу з урахуванням факторів ризику, що входять до Фремінгемської шкали, погіршену вазодилатуючу функцію було визнано незалежним предиктором вищезазначених подій [20]. Схожі дані отримано і в Health Study серед подібної когорти осіб (n = 2264) [21]. Водночас у дослідженнях FATE та PIVUS такий зв’язок не виявлено, що може бути пов’язано з певними методологічними особливостями. Так, у FATE встановлено зв’язок зниженої ЕЗВД із мікроваскулярною дисфункцією судин міокарда, що, у свою чергу, корелювала з довготерміновим прогнозом, а в PIVUS негативний результат пов’язують із включенням великої кількості молодих осіб [22].
У роботах [23, 24] ЕД КА у 157 хворих виявилася незалежним предиктором кардіоваскулярних подій (ССС, ІМ). У роботі [25] було продемонстровано, що знижена ЕЗВД, визначена у пробі з РГ, має таке саме прогностичне значення щодо виникнення серцево-судинних подій у хворих на ішемічну хворобу серця (ІХС), як і дисфункція КА у пробі з АХ. У низці робіт таке ж її значення було виявлено й у пацієнтів з атеросклерозом периферичних артерій [26, 27].
Схожі дані отримано у хворих із нестабільним перебігом ІХС. Так, Elbaz і співавт. серед хворих із гострим коронарним синдромом (ГКС) майже у всіх виявили ЕД КА, що зберігалася і протягом наступних місяців після включення в дослідження [28]. О.М. Пархоменко і співавт. визначили, що погіршена проба з РГ у хворих на ГКС асоціюється з розвитком ранньої –післяінфарктної стенокардії та рецидивом ІМ [84]. У дослідженнях [29, 30] ЕД, що визначалася у пробі з РГ у хворих після перенесеного ГКС, була предиктором повторних серцево-судинних подій, і навпаки, у разі нормалізації приросту діаметра плечової артерії (ПА) у таких пацієнтів зменшувався ризик зазначених подій.
Для хронічної систолічної серцевої недостатності як ішемічного, так і ідіопатичного генезу характерна ЕД. Прогностична її значущість на сьогодні добре вивчена. Уперше зв’язок ЕД при хронічній серцевій недостатності (ХСН) із систолічною дисфункцією лівого шлуночка (СД ЛШ) і виживанням був досліджений Fisher і спів–авт. [31]. Під час аналізу виживання хворих протягом 45,7 мiс. залежно вiд величини приросту діаметра ПА у фазу РГ виявлено, що нижчі його значення асоцiйованi з достовiрно менш сприятливим прогнозом життя, ніж у хворих із більшим показником (рис. 1).
/71.jpg)
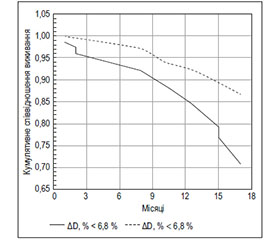
Раніше нами виявлено, що 12-місячна виживаність хворих на ХСН і СД ЛШ (n = 162) залежала від величини приросту діаметра ПА у фазу РГ. Предиктором достовірно гіршого виживання протягом року була величина приросту ПА менше за 6,8 % (рис. 2) [32]. В обох зазначених дослідженнях групи хворих, що були розподілені за медіаною приросту ПА у фазу РГ, мали порівнянний показник фракції викиду (ФВ) ЛШ, що дозволило авторам дійти висновку про самостійну патогенетичну роль ЕД у прогресуванні ХСН.
Так, S.D. Katz і співавт. виявили серед 259 хворих ХСН і СД ЛШ (дилатаційна кардіоміопатія (ДКМП) або ІХС) гірший прогноз (смерть або необхідність трансплантації серця) виживання протягом 2,3 року спостереження в тих, хто мав нижчі значення приросту діаметра ПА у фазу РГ. У роботі також було виявлено асоціацію гіршого прогнозу виживання з меншою концентрацією NO у видихуваному повітрі після навантажувального тесту [33].
У дослідженні, подібному за дизайном до вищеза-значених, B. Meyer і співавт. серед 75 хворих з ХСН на тлі оптимізованого передбаченого стандартами лікування спостерігали у пацієнтів з ЕЗВД < 6,8 % (медіана) гірші показники довготривалого виживання (3 роки). Після проведеного багатофакторного аналізу, окрім низьких значень ЕЗВД, із гіршим прогнозом виживання асоціювався підвищений рівень попередника мозкового натрійуретичного гормона [34].
Протягом 14 міс. спостереження M. Shechter і спів-авт. встановили серед хворих з ХСН ІV функціонального класу (ФК) за NYHA (n = 82) ішемічного генезу зв’язок гіршого прогнозу (смерть, повторна госпіталізація з приводу декомпенсації ХСН або ІМ) з низьким значенням ЕЗВД (≤ 4,6 %), визначеної у пробі з РГ. У цих хворих також виявили достовірно гіршу ендотелійнезалежну вазодилатацію (проба з нітрогліцерином). Ураховуючи отримані результати, автори зазначають, що ЕД у такої категорії хворих поряд з іншими чинниками може бути зумовлена також і структурними змінами в судинній стінці [35]. У роботі [36] негативне прогностичне значення потікзалежної вазодилатації серед 242 хворих з ХСН було визначено із застосуванням венооклюзивної плетизмографії.
Крім того, J. Akar і співавт. встановили серед хворих з ХСН і СД ЛШ прогностичну значущість ЕЗВД, виміряну у пробі з РГ, щодо кращої відповіді на тлі застосування ресинхронізуючої терапії (CRT) [37]. В інших двох дослідженнях [38, 39] показано, що призначення CRT покращує вазодилатуючу функцію ендотелію. Дослідники припускають, що отриманий результат може бути опосередкований зміною напруги зсуву внаслідок покращення насосної функції серця.
Розглянемо основні чинники зниження біодоступності NO при ХСН. Так, кількість NO зменшується, з одного боку, у результаті пригнічення стимуляції ендотеліальної NO-синтази, з іншого — унаслідок збільшення його інактивації, переважно вільними радикалами кисню (О2–). Одним із важливих чинників зменшення стимуляції eNOS є зміна напруги зсуву (shear stress), що виникає через зниження швидкості системного кровообігу, характерного для ХСН [40, 41]. Під напругою зсуву розуміють силу гідродинамічної дії потоку крові на клітини ендотелію. Зазначена сила активує калієві канали ендотеліоцитів і зумовлює підвищення в них концентрації іонів кальцію, які активують еNOS, стимулюють синтез нею NO, під їх впливом також збільшується синтез простацикліну та пригнічується синтез ендотеліну-1 [42].
Інший чинник, що здатний пригнічувати eNOS, — характерна для ХСН гіперактивація ренін-ангіотензинової системи (РАС), зокрема властивість ендотеліальної АПФ прискорювати розпад брадикініну з розвитком його відносного дефіциту. Відсутність адекватної стимуляції брадикінінових В2-рецепторів ендотеліоцитів призводить до зниження активації ним ферменту eNOS і, відповідно, синтезу NO, що зумовлює підвищення тонусу гладком’язових клітин судинної стінки [44]. Вагоме значення брадикініну щодо впливу на тонус судин було показане в дослідженні за участю хворих, які мали гіпоренінову форму ГХ. На фоні прийому інгібітору АПФ (ІАПФ) у них спостерігалося достовірне зниження артеріального тиску, спричинене саме зменшенням прискореного розпаду брадикініну [45].
Підвищений рівень А ІІ не лише зумовлює вазоконстрикцію, а й має прооксидантну дію, що реалізується шляхом активації НАДФН-оксидаз. Останні здатні окислювати НАДФ+ із утворенням супероксиду кисню. В експериментальних роботах показано, що частково зазначений механізм реалізується через тканинну альдостеронову систему [46]. Так, у роботі J. Bauersachs і співавт. під дією еплеренону знижувалася продукція вільних радикалів, що зумовлено зниженням синтезу субодиниці p22phox НАДФН-оксидази [47]. У свою чергу, вільні радикали кисню вступають у реакцію з NO, нейтралізуючи його позитивні ефекти, та утворюють метаболіт — перекиснітрит [48]. Окрім зазначеного, А ІІ стимулює експресію факторів росту та розвиток фіброзу в судинній стінці [44].
Наступний механізм зниження біодоступності NO — підвищення рiвня прозапальних цитокiнiв і TNF, що пригнічують синтез NO [49]. В експериментальних дослідженнях доведено, що TNF пригнічує вивільнення NO з ендотеліальних клітин [50] та скорочує період напівжиття матричної рибонуклеїнової кислоти гена eNOS [51]. Відомо також, що TNF може стимулювати синтез вільних радикалів гладком’язовими клітинами судинної стінки, що призводить до інактивації NO [52].
Іншим вагомим джерелом вільних радикалів є ксантиноксидаза, що каталізує окислення ксантину та гіпоксантину. Виявлено підвищення активності ксантиноксидази в мишей із модельованою СН, тоді як внутрішньовенне введення алопуринолу покращувало ендотеліальну дисфункцію [53]. Окрім цього, низький рівень NO сам по собі може потенціювати оксидантний стрес. У дослідах показано, що активність супероксиддисмутази (СОД), одного з найпотужніших антиоксидантних ферментів, який каталізує перетворення супероксиду на кисень і перекис водню, залежить від рівня NO. У разі зменшення останнього активність СОД знижується, що може призводити до збільшення кількості вільних радикалів кисню [54]. Необхідно також зазначити, що швидкість взаємодії останнього з NO втричі вища, ніж із СОД [8].
Зниження синтезу NO може зумовлюватися також зміною активності та/або кількості ферменту ендотеліальної NO-синтази (eNOS) внаслідок поліморфізму гена, що кодує її. Генетичний поліморфізм характеризується наявністю більше двох варіантів алелей у популяції. Зазвичай різниця між алелями одного і того ж гена незначна, вона полягає у заміні одного нуклео-тиду або зміні числа повторюваних фрагментів ДНК. Ген eNOS розташований на довгому плечі 7-ї хромосоми (7q35-36) і складається з 26 екзонів і 25 інтронів; загальна кількість пар нуклеотидів — близько 21 тис. Функціональне значення та зв’язок із серцево-судинними захворюваннями найбільш вивчений для трьох поліморфних варіантів гена eNOS: поліморфізм промотора Т(-786)С, 7 екзона G894Т та четвертого інтрона (4b/4a).
Промоторна ділянка гена eNOS містить сайти для зв’язування з транскрипційними факторами (активуючий протеїн 1 та 2), має естроген-чутливий елемент і елемент, що реагує на напруження зсуву. Серед шести поліморфних варіантів промотора найбільше функціональне значення встановлено для Т(-786)С [55, 56]. M. Nakayama і співавт. за допомогою люциферазного тесту довели, що в разі заміни тиміну (Т) на цитозин (С) в положенні (-786) промотора його активність знижується, тоді як поліморфізм інших ділянок промотора не змінював його активності [56]. Припускають, що можливий механізм впливу мутації Т(-786)С у промоторі на зчитування гена eNOS зумовлений специфічним зв’язуванням білка реплікації А1 зі зміненим сайтом промотора [57]. Цей протеїн відомий як білок, що має здатність до зв’язування з одноланцюговими молекулами ДНК і є необхідним для репарації, реплікації та рекомбінації. Саме за рахунок зв’язування з білком реплікації А1 знижується активність промотора в разі заміни тиміну на цитозин (Т(-786)С). Це підтверджується тим, що введення олігонуклеотидної послідовності, комплементарної до згаданого білка, відновлює транскрипційну активність промотора гена eNOS за наявності зазначеного поліморфізму. У разі зниженої активності промотора внаслідок заміни Т(-786)С спостерігається зменшення кількості інформаційних РНК eNOS і зменшення кількості білкових молекул eNOS, що в подальшому призводить до відповідного зменшення синтезу NO. A. Doshi і співавт., застосовуючи кількісну полімеразну ланцюгову реакцію, під час вивчення зразків міокарда хворих з ХСН відмітили достовірно меншу кількість матричної РНК білка eNOS у гомозигот СС порівняно з носіями алелі Т (гомозиготи ТТ і гетерозиготи ТС). Експресія білка eNOS також була меншою в носіїв СС-генотипу [58]. Такі дані підтверджуються й у дослідах на культурі клітин людини. Експресія ферменту eNOS у носіїв генотипу СС поліморфізму Т(-786)С під дією моделі, подібної за механізмом дії до напруження зсуву, була меншою, ніж у гомозигот ТТ [59].
Поліморфізм G894Т 7 екзона характеризується заміною азотистої основи гуанін (G) на тимін (Т), що призводить до заміни глутамінової амінокислоти (Glu) на аспарагінову (Asp) у положенні 298 білка eNOS (Glu298Asp). Заміна амінокислоти відбувається в оксигеназному домені ферменту eNOS, який не відповідає за його активність. Можливо, тому в більшості досліджень, що мали на меті визначити функціональне значення цього ферменту, не встановлено різниці в активності білка eNOS серед носіїв генотипів GG, GT і ТТ. Так, у вже згадуваному дослідженні A. Doshi не було виявлено достовірної різниці в кількості матричної РНК eNOS та експресії білка eNOS у носіїв рідкісної алелі Т (гомозиготи ТТ та гетерозиготи GT) порівняно з гомозиготами GG [58]. У роботі R. Golser і співавт. було встановлено, що мутантний варіант білка не поступається ні за афінністю до L-аргініну, ні за інтенсивністю утворення цитруліну, оксидоредуктазною активністю, чутливістю до кальцію, кальмодулінзв’язуючою активністю та іншими властивостями [84]. Проте відсутність відмінностей ізольованого білка не обов’язково свідчить про те, що аналогічна закономірність спостерігатиметься в живих клітинах, де активність eNOS у кавеолах цитоплазматичної мембрани визначається оточуючими умовами та контролюється багатьма іншими білками. Так, у роботі M. Tesauro і співавт. було визначено, що білок із заміною Glu на Asp у положенні 298 легко розщеплюється на два фрагменти: 35 кДа N-термінальний і 100 кДа С-термінальний [60]. Гідроліз відбувається саме за зв’язком, що утворюється між Asp298 і Pro299. За умов ex vivo це розщеплення потенціюється підвищенням температури та зниженням рівня рН. Передбачається, що це розщеплення є ацидотичним гідролізом білка eNOS невідомою протеїназою [61]. У дослідженні T.A. Fairchild і співавт. було показано, що в культурі клітин COS7, трансфектованих ДНК нормального гена eNOS і гена із поліморфізмом G894Т, різниці в рівні виділення в середовище метаболітів NO не було, тобто як нормальний, так і мутантний білок є каталітично активними та забезпечують нормальний синтез NO за звичайних умов культивування. Однак у разі зменшення рН середовища нижче від 5,0 упродовж 10 год відбувалось утворення 100 кДа фрагмента та зменшення періоду напівжиття мутантної форми ензиму. Застосування різноманітних екстремальних факторів, зокрема впливу гіпоксичної суміші протягом 48 год, відтворення оксидантного стресу за допомогою перекису водню або вплив цитостатика стауроспорину, не призводило до фрагментації мутантного варіанта eNOS. Автори не виключають, що феномен ацидотичного гідролізу G894Т варіанта eNOS є артефактом, генерованим in vitro, однак із нормальним білком відповідних змін не відбувалося. У будь-якому разі є підстави вважати, що алельний поліморфізм G894Т збільшує вразливість білка до гідролізу, особливо за умови дії патологічних чинників. Унаслідок цього вміст білка зменшується, що зумовлює розвиток патофізіологічних проявів недостатності ендогенного синтезу NO [61].
Поліморфізм 4-го інтрона 4b/а полягає у варіабельній кількості тандемних повторів 27 нуклеотидів. Кількість останніх може становити два (алель 4у), чотири (4а), п’ять (4b) та шість (4с). Алельні варіанти 4у і 4с зустрічаються вкрай рідко лише серед негроїдної раси. Відомо, що інтрони не несуть інформації про амінокислотну послідовність білка та вирізаються під час сплайсингу РНК. Є дані, що за наявності алелі 4а порушується процес транскрипції матричної РНК ферменту eNOS [61].
З часу визначення поліморфізмів гена eNOS почалося активне вивчення їх клінічного значення в здорових осіб, хворих на ГХ, ІХС. Більшість дослідників, проте не всі [62], визначають вплив поліморфізму промотора Т(-786)С у здорових осіб на вазодилатуючу функцію ендотелію (гірша ЕЗВД визначена у пробі з РГ [63] під час вивчення концентрації нітритів у крові [64]). Такі неоднозначні дані були отримані серед хворих на ГХ [65, 66], ІХС [67], ГКС [68]. Під час дослідження поліморфізму 7-го екзона G894Т гена eNOS серед волонтерів було виявлено гіршу ЕЗВД у пробі з АХ у носіїв рідкісного генотипу ТТ порівняно з GG [65]. Схожі результати були отримані і серед хворих на ГХ (n = 51) [70], тоді як в іншій роботі при порівнянні 235 пацієнтів із ГХ і 94 здорових осіб різниці в ЕЗВД встановлено не було [65]. Отже, дані щодо впливу поліморфізмів гена eNOS на вазодилатуючу функцію ендотелію серед здорових осіб, хворих на ГХ, ІХС, суперечливі. Такий зв’язок у хворих із ХСН вивчався в роботі M. Kose і співавт., які виявили, що у хворих (n = 104) — носіїв алелі а поліморфізму 4b/а гена eNOS та алелі С поліморфізму AT1R — ЕЗВД була гіршою, тоді як поліморфізм АПФ (I/D) впливу не мав [71].
Ми досліджували вазодилатуючу функцію ендотелію залежно від поліморфізму промотора Т(-786)С серед хворих із ХСН і СД ЛШ (середня ФВ ЛШ 35,0 [22,0; 42,5] %) (n = 116). У всіх хворих ЕЗВД, оцінена в пробі з РГ, була достовірно меншою порівняно з такою у контрольній групі, у якій приріст ПА становив 12,5 [10,2; 14,3] %. Серед гомозигот ТТ поліморфізму Т(-786)С гена eNOS показник ЕЗВД становив 7,2 [4,9; 8,3] %, у гетерозигот ТС — 6,6 [4,4; 9,1]%, тоді як в осіб з рідкісним генотипом СС — 4,7 [2,8; 6,0] %. В останніх ЕЗВД є достовірно меншою порівняно з такою у носіїв алелі Т (ТТ, ТС) (рис. 3).
Водночас хворі з генотипами ТТ, ТС та СС не відрізнялися за основними клініко-демографічними показниками (віком, давністю артеріальної гіпертензії, ІХС, кількістю перенесених ІМ, ФК за NYHA, рівнем артеріального тиску і частотою серцевих скорочень), морфофункціональними змінами міокарда (індекс кінцево-систолічного, кінцево-діастолічного об’єму, ФВ, індекс маси міокарда, розмір лівого передсердя і рівень систолічного тиску в легеневій артерії). Оскільки ступінь вираженості системного оксидантного стресу й активність антиоксидантних ферментів можуть впливати на біодоступність NO та, відповідно, на стан ЕЗВД, ми досліджували показники перекисного окислення (малоновий діальдегід, дієнові кон’югати) та активність ензимів антиоксидантного захисту (СОД, глутатіонредуктази) у хворих з різними варіантами поліморфізму Т(-786)С гена eNOS. За рівнем зазначених величин групи не відрізнялися. Лікування, зокрема прийом ІАПФ і статинів, також впливає на ЕЗВД, проте в досліджуваних групах дози зазначених лікарських засобів не відрізнялися. Хворі, які приймали карведилол чи небіволол (що, як відомо, можуть покращувати біодоступність NO), у дослідження не включалися.
В осіб з рідкісним генотипом ТТ поліморфізму 7 екзона G894T гена еNOS також відмічено достовірно гіршу ЕЗВД — 4,2 [2,5; 5,3] % порівняно з гетерозиготами GТ — 6,2 [5,1; 8,1] %, та гомозиготами GG — 7,1 [4,3; 9,4] % (рис. 4). За основними клініко-інструментальними даними, лабораторними показниками та лікуванням, що зазначені вище, хворі трьох генотипів поліморфізму G894T гена еNOS не відрізнялися.
Під час вивчення комбінацій генотипів обох досліджуваних поліморфізмів було виявлено, що найгірші значення вазодилатуючої функції ендотелію мали хворі з гомозиготним станом рідкісних алелей: СС/ТТ (n = 8), ЕЗВД у них становила 4,1 [2,4; 5,0] % [72].
Оскільки прогностичне значення вазодилатуючої функції ендотелію доведено в багатьох дослідженнях [31–36], постає запитання: чи можуть поліморфізми гена eNOS впливати на частоту госпіталізації з приводу серцево-судинних подій, кардіальну чи судинну смертність? Такий зв’язок широко вивчений серед хворих на ІХС і лише в декількох роботах у пацієнтів із ХСН. Так, у дослідженні GENICA study вивчали поліморфізми Т(-786)С та G894Т гена eNOS у 1086 пацієнтів із коронарним атеросклерозом [73]. Після 3,5 року спостереження виявлено збільшення серцево-судинної смерті в носіїв СС-генотипу Т(-786)С і відсутність різниці між генотипами поліморфізму G894Т гена eNOS. T. Nishijima і співавт. після 5 років спостереження (n = 201) встановили більшу частоту повторного коронарного спазму в носіїв алелі С поліморфізму Т(-786)С [74], тоді як серед населення Ірану (n = 207) визначено збільшення ризику виникнення ІХС в 2,1 раза в носіїв алелі Т поліморфізму G894Т гена eNOS [75]. Метааналіз (2014 р.) 24 досліджень (6192 хворі на ІХС і 9281 здорова особа) підтвердив зв’язок поліморфізму Т(-786)С гена eNOS із ризиком виникнення ІХС [76].
У хворих з ХСН і СД ЛШ (n = 469), серед яких 51 % становили особи із ДКМП, D. McNamara і співавт. визначили асоціацію алелі Т поліморфізму G894Т гена eNOS з гіршим прогнозом (ССС або потреба в трансплантації комплексу «серце — легені»). Особливо тісний зв’язок спостерігався для пацієнтів із ХСН неішемічного генезу [77]. K. Kim і співавт. досліджували вплив шести генів-кандидатів (α(2C)-AR (ADRA2C-), β1-AR (ADRB1), β2-AR (ADRB2), eNOS, АПФ (I/D) та CYP4A11) на частоту повторних госпіталізацій з приводу декомпенсації ХСН (n = 140). З більшим ризиком госпіталізації асоціювалися гомозиготи ТТ поліморфізму G894Т гена eNOS, а також носії алелі Arg генотипу Gly389Arg ADRB1 та гомозиготи Gly16Gly поліморфізму Arg16Gly ADRB2, тоді як інші досліджувані поліморфізми не впливали на прогноз [78].
Водночас у бразильській популяції (n = 145) хворих із ХСН і СД ЛШ не було виявлено впливу поліморфізму G894Т гена eNOS на виживання та частоту госпіталізацій з приводу ХСН [79]. N. Martinelli і співавт. серед подібної популяції (n = 145) визначили протективний щодо прогнозу та госпіталізації з приводу ХСН гаплотип 786C/4b/Asp298 [80]. Такі результати у вищезазначених двох дослідженнях можуть бути пов’язані з переважанням серед їх учасників пацієнтів негроїдної раси.
У нашій роботі [81] ми досліджували поліморфізми Т(-786)С та G894Т гена eNOS серед хворих із ХСН і СД ЛШ (n = 104) протягом 2,5 року. У гомозигот СС (n = 19) виявлено гірший прогноз як щодо госпіталізації з приводу декомпенсації ХСН (р = 0,015), ССС (р = 0,046), так і щодо комбінованої події (р = 0,010) (рис. 5) порівняно з гомозиготами ТТ (n = 40).
Під час аналізу прогнозу серед носіїв різних генотипів поліморфізму G894Т з огляду на невелику чисельність гомозигот за рідкісною алеллю Т їх об’єднали з гетерозиготами GT + TT (n = 45) та порівнювали з гомозиготами GG (n = 59). У результаті такого аналізу не було виявлено різниці в прогнозі. Зазначимо, що за умови достатньої кількості спостережень серед носіїв рідкісного генотипу ТТ і порівняння цієї групи окремо з гомозиготами GG, можливо, результати могли б бути більш статистично значущими [82].
Варто зауважити, що в порівнюваних групах хворих ми аналізували прогнозмодулюючі фактори (прийом і дози бета-блокаторів, ІАПФ, антагоністів мінералокортикоїдних рецепторів, антитромбоцитарних препаратів і/або антикоагулянтів; стентування КА, аортокоронарне шунтування). За наведеними показниками групи не відрізнялися на момент включення в дослідження та через 2,5 року.
Отже, є підстави розглядати алельний поліморфізм промотора Т(-786)С гена eNOS як один із чинників, що асоційований зі станом ендотеліальної функції та довготерміновим клінічним прогнозом пацієнтів із ХСН. Попри достовірний зв’язок G894Т поліморфізму гена eNOS зі станом вазодилатуючої функції ендотелію його роль як чинника довготермінового прогнозу серед таких пацієнтів потребує подальшого уточнення. Іншим важливим питанням залишається те, наскільки ці поліморфізми можуть обмежувати фармакодинамічну відповідь лікарських засобів, механізм дії яких полягає в стимуляції ендотеліальної NO-синтази.
Список литературы
1. Remzi Onder M., Barcu Barutcuogu. The endothelium. — 1999. — P. 55.
2. Беленков Ю.Н. Эндотелиальная дисфункция при сердечной недостаточности: возможности терапии ингибиторами ангиотензинпревращающего фермента / Ю.Н. Беленков, В.Ю. Мареев, Ф.Т. Агеев // Кардиология. — 2001. — № 5. — С. 100-104.
3. Cloning and structural characterization of the human endothelial nitric-oxide-synthase gene / K. Miyahara, T. Kawamoto, K. Sase [et al.] // Eur. J. Biochem. — 1994. — Vol. 223 (3). — P. 719-726.
4. Vanhoutte P. et al. Endothelial dysfunction and vascular disease. The endothelium and clinical practice / Ed. by Gabor M. Rubanyi, Victor J. Dzau. — NY, 1997.
5. Branwuald E. Heart disease. — 5th ed. — Suanders, Philadelphia, 1997.
6. Vane J. Regulatiry function of the vascular endothelium / J. Vane, E. Anggard, R. Botting // N. Engl. J. Med. — 1999. — Vol. 323. — P. 27-36.
7. Mechanisms of nitric oxide release from the vascular endothelium / R. Busse, A. Mulsch, I. Fleming [et al.] // Circulation. — 1993. — Vol. 87. — P. 18-25.
8. Vane J. Regulatiry function of the vascular endothelium / J. Vane, E. Anggard, R. Botting // N. Engl. J. Med. — 1999. — Vol. 323. — P. 27-36.
9. Dulak J. Nitric oxide induces the synthesis of vascular endothelial growth factor / J. Dulak, A. Jozkowicz, A. Dembinska-Kiec // Ateroscler. Thromb. Vasc. Biol. — 2000. — Vol. 20. — P. 659-666.
10. Cooce J. Nitric oxide and angiogenesis / J. Cooce, D. Lo-sordo // Circulation. — 2002. — Vol. 105. — P. 81-96.
11. Amiya E., Watanabe M., Komuro I. The Relationship between Vascular Function and the Autonomic Nervous System // Ann. Vasc. Dis. — 2014. — Vol. (2). — P. 109-119.
12. D’Apolito M., Du X., Pisanelli D., Pettoello-Mantovani M., Campanozzi A., Giacco F., Maffione A.B., Colia A.L., Brownlee M., Giardino I. Urea-induced ROS cause endothelial dysfunction in chronic renal failure // Atherosclerosis. — 2015. — Vol. 239 (2). — P. 393-400.
13. Urban M.H., Valipour A., Kiss D., Eickhoff P., Funk G.C., Burghuber O.C. Soluble receptor of advanced glycation end-products and endothelial dysfunction in COPD // Respir. Med. — 2014. — Vol. 108 (6). — P. 891-897.
14. Lekakis J., Abraham P., Balbarini A., Blann A., Boulanger C.M., Cockcroft J., Cosentino F., Deanfield J., Gallino A., Ikonomidis I., Kremastinos D., Landmesser U., Protogerou A., Stefanadis C., Tousoulis D., Vassalli G., Vink H., Werner N., Wilkinson I., Vlachopoulos C. Methods for evaluating endothelial function: a position statement from the European Society of Cardiology Working Group on Peripheral Circulation // Eur. J. Cardiovasc. Prev. Rehabil. — 2011. — Vol. 18. — P. 775-789.
15. Panza J. et al. Role of endothelium-derived in the abnormal endothelium — dependent vascular relaxation of patient with essential hypertension // Circulation. — 1993. — Vol. 87. — P. 1468-1474.
16. Taddei S., Virdis A., Mattei P., Ghiadoni L., Fasolo C.B., Sudano I., Salvetti A. Hypertension causes premature aging of endothelial function in humans // Hypertension. — 1997. — Vol. 29 (3). — P. 736-743.
17. Yasue H.I., Nakagawa H., Itoh T., Harada E., Mizuno Y. Coronary artery spasm — clinical features, diagnosis, pathogenesis, and treatment // J. Cardiol. — 2008. — Vol. 1 (1). — P. 2-17.
18. Rossi R., Nuzzo A., Origliani G., Modena M.G. Prognostic role of flow-mediated dilation and cardiac risk factors in post-menopausal women // J. Am. Coll. Cardiol. — 2008. — Vol. 51 (10). — P. 997-1002.
19. Suzuki T., Hirata K., Elkind M.S., Jin Z., Rundek T., Miyake Y., Boden-Albala B., Di Tullio M.R., Sacco R., Homma S. Me–tabolic syndrome, endothelial dysfunction, and risk of cardiovascular events: the Northern Manhattan Study (NOMAS) // Am. Heart J. — 2008. — Vol. 156. — P. 405-410.
20. Yeboah J., Folsom A.R., Burke G.L., Johnson C., Polak J.F., Post W., Lima J.A., Crouse J.R., Herrington D.M. Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: the multi-ethnic study of atherosclerosis // Circulation. — 2009. — Vol. 120. — P. 502-509.
21. Yeboah J., Crouse J.R., Hsu F.C., Burke G.L., Herrington D.M. Brachial flow-mediated dilation predicts incident cardiovascular events in older adults: the Cardiovascular Health Study // Circulation. — 2007. — Vol. 115. — P. 2390-2397.
22. Enrique Gutiérrez, Andreas J. Flammer, Lilach O. Lerman, Jaime Elízaga, Amir Lerman, Francisco Fernandez-Aviles. Endothelial dysfunction over the course of coronary artery disease // Eur. Heart J. — 2013. — Vol. 34 (41). — P. 3175-3181.
23. Suwaidi J.A., Hamasaki S., Higano S.T., Nishimura R.A., Holmes D.R. Jr, Lerman A. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction // Circulation. — 2000. — Vol. 101. — P. 948-954.
24. Schachinger V., Britten M.B., Zeiher A.M. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease // Circulation. — 2000. — Vol. 1. — P. 1899-1906.
25. Heitzer T., Schlinzig T., Krohn K., Meinertz T., Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease // Circulation. — 2001. — Vol. 104. — P. 2673-2678.
26. Сharakida M. Asseeement of atherosclerosis: the role of flow-mediated dilatation // Eur. Heart J. — 2010. — Vol. 31. — P. 2854-2861.
27. Brevetti G., Schiano V., Chiariello M. Endothelial dysfunction: a key to the pathophysiology and natural history of peripheral arterial disease? // Atherosclerosis. — 2008. — Vol. 197 (1). — P. 1-11.
28. Elbaz M., Carrie D., Baudeux J.L., Arnal J.F., Maupas E., Lotterie J.A., Perret B., Puel J. High frequency of endothelial vasomotor dysfunction after acute coronary syndromes in non-culprit and angiographically normal coronary arteries: a reversible phenomenon // Atherosclerosis. — 2005. — Vol. 181. — P. 311-319.
29. Fichtlscherer S., Breuer S., Zeiher A.M. Prognostic value of systemic endothelial dysfunction in patients with acute coronary syndromes: further evidence for the existence of the «vulnerable» patient // Circulation. — 2004. — Vol. 110. — P. 1926-1932.
30. Careri G., Nerla R., Di Monaco A., Russo G., Stazi A., Villano A., Sestito A., Lanza G.A., Crea F. Clinical correlates and prognostic value of flow mediated dilation in patients with non-ST segment elevation acute coronary syndromes // Am. J. Cardiol. — 2013. — Vol. 111. — P. 51-57.
31. Fischer D. Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation or death / D. Fischer, S. Rossa, U. Landmesser // Europ. Heart J. — 2005. — Vol. 26. — P. 65-69.
32. Воронков Л.Г. Ендотелійзалежна вазодилятація та її прогностичне значення у хворих хронічною серцевою недостатністю і систолічною дисфункцією лівого шлуночка / Л.Г. Воронков, І.А. Шкурат, Є.М. Бесага // Український кардіологічний журнал. — 2005. — № 6. — С. 23-30.
33. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure / S.D. Katz, K. Hryniewicz, I. Hriljac, K. Balidemaj, C. Dimayuga, A. Hudaihed, A. Yasskiy // Circulation. — 2005. — Vol. 111 (3). — P. 310-314.
34. Flow-mediated vasodilation predicts outcome in patients with chronic heart failure: comparison with B-type natriuretic peptide / B. Meyer, D. Mortl, K. Strecker, M. Hulsmann, V. Kulemann, T. Neunteufl, R. Pacher, R. Berger // J. Am. Coll. Cardiol. — 2005. — Vol. 46 (6). — P. 1011-1018.
35. Long-term association of brachial artery flow-mediated vasodilation and cardiovascular events in middle-aged subjects with no apparent heart disease / M. Shechter, A. Issachar, I. Marai, N. Koren-Morag, D. Freinark, Y. Shahar, A. Shechter, M. S. Feinberg // Int. J Cardiol. — 2009. — Vol. 134 (1). — P. 52-58.
36. Berrazueta R. Endothelial dysfunction, measured by reactive hyperaemia using strain-gauge plethysmography, is an independent predictor of adverse outcome in heart failure / R. Berrazueta, A. Guerra-Ruiz, T. Garcia-Unzueta // J. Heart Fail. — 2010. — Vol. 12 (5). — P. 477-483.
37. Akar J.G., Al-Chekakie M.O., Fugate T., Moran L., Froloshki B., Varma N., Santucci P., Wilber D.J., Matsumura M.E. Endothelial dysfunction in heart failure identifies responders to cardiac resynchronization therapy // Heart Rhythm. — 2008. — Vol. 5. — P. 1229-1235.
38. Enomoto K., Yamabe H., Toyama K., Matsuzawa Y., Yamamuro M., Uemura T., Morihisa K., Iwashita S., Kaikita K., Sugiyama S., Ogawa H. Improvement effect on endothelial function in patients with congestive heart failure treated with cardiac resynchronization therapy // J. Cardiol. — 2011. — Vol. 8. — P. 69-73.
39. Tesselaar E., Schiffer A., Widdershoven J., Broers H., Hendriks E., Luijten K., Creusen J. Effect of cardiac resynchronization therapy on endothelium-dependent vasodilatation in the cutaneous microvasculature // Pacing Clin. Electrophysiol. — 2012. — Vol. 35. — P. 377-384.
40. Harrison D., Widder J. Endothelial mechanotransduction, nitric oxide and vascular inflammation // J. Intern. Med. — 2006. — Vol. 259. — P. 351-363.
41. Zelis R. A comparison of the effect of vasodilator stimuli on peripheral resistence vessels in normal subject and in patients with congestive heart failure / R. Zelis, D. Mason, E. Braunwald // J. Clin. Invest. — 1998. — Vol. 47. — P. 960-970.
42. Increased levels of inflammatory cytokines and endothelin-1 in alveolar macrophages from patients with chronic heart failure / L.I. Sikkeland, C.P. Dahl, T. Ueland, A.K. Andreassen, E. Gude, T. Edvardsen, T. Holm, A. Yndestad, L. Gullestad, J. Kongerud, P. Aukrust // Nitric Oxide. — 2012. — Vol. 26 (3). — P. 141-147.
43. Angiotensin-converting enzyme insertion/deletion polymorphism modulates the human in vivo metabolism of bradykinin / L.J. Murphey, J.V. Gainer, D.E. Vaughan [et al.] // Circulation. — 2000. — Vol. 102. — P. 829-832.
44. Aldosterone deficiency and mineralocorticoid receptor antagonism prevent angiotensin II-induced cardiac, renal, and vascular injury / J.M. Luther, P. Luo, Z. Wang, S.E. Cohen, H.S. Kim, A.B. Fogo, N.J. Brown // Kidney Int. — 2012. — Vol. 23. — P. 34-39.
45. Hornig C. Role of bradykinin in mediating vascular effects of ACE inhibitors in humans / C. Hornig, C. Kohler, H. Drexler // Circulation. — 1997. — Vol. 95. — P. 1115-1118.
46. Widder J., Behr T. et al. Vascular endothelial dysfunction and superoxide anion production in heart failure are p38 MAPkinase-dependent // Cardiovasc. Res. — 2004. — Vol. 63. — P. 161-167.
47. Bauersachs J. et al. Endothelial dysfunction in chronic myocardial infarction despite increased vascular Endothelial nitric oxide synthase and solube guanylate cyclase expression: role of enhanced vascular superoxide production // Circulation. — 1999. — Vol. 100. — P. 292-298.
48. Beckman J.S. Nitric oxide reversibly inhibits Fas-induced apoptosis / J.S. Beckman, W.H. Koppenol // J. Biol. Chem. — 1997. — Vol. 272. — P. 240-245.
49. Katz S.D. Pathophysiological correlation of increased serum tumor necrosis factor in patient with chronic heart failure. Relation to nitric oxide dependent vasodilatation in the forearm circulation / S.D. Katz, R. Ramanath // Circulation. — 1994. — Vol. 90. — P. 12-16.
50. Elevated circulation levels of tumor necrosis factor in severe chronic heart failure / B. Levine, J. Kalman, L. Mayer [et al.] // N. Engl. J Med. — 1990. — Vol. 323. — P. 236-241.
51. Yoshimuzi M. Tumor necrosis factor dowregulates an endothelial nitric oxide synthase mRNC by shortening its half-life / M. Yoshi- muzi, M. Perella, J. Burnett // Circ. Res. — 1993. — Vol. 73. — P. 205-209.
52. Matsabura T. Increased superoxide anioin release from human endothelial cells in response to cytokines / T. Matsabura, M. Ziff // J. Immunol. — 1986. — Vol. 137. — P. 3295-3298.
53. de Jong J.W., Schoemaker R. et al. Enhanced expression and activity of xanthine oxidoreductase in the failing heart // J. Mol. Cell Cardiol. — 2000. — Vol. 32. — P. 2083-2089.
54. Comperative effect of ACE inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease. Role of superoxide dismutase / B. Hor–nig, U. Landmesser, C. Kohler [et al.] // Circulation. — 2001. — Vol. 104. — P. 2177-2181.
55. Wang J. Haplotype-specific effects on endothelial NO synthase promoter efficiency: modifiable by cigarette smoking / J. Wang, D. Dudley, X. L. Wang // Arterioscler. Thromb. Vasc. Biol. — 2002. — Vol. 5. — P. 1-4.
56. Synergistic interaction of T-786->C polymorphism in the endothelial nitric oxide synthase gene and smoking for an enhanced risk for coronary spasm / M. Nakayama, M. Yoshimura, T. Sakamoto [et al.] // Pharmacogenetics. — 2003. — Vol. 11. — P. 683-688.
57. Replication protein A1 reduces transcription of the endothelial nitric oxide synthase gene containing a-786T->C mutation associated with coronary spastic angina / Y. Miyamoto, Y. Saito, M. Nakayama [et al.] // Hum. Mol. Genet. — 2000. — Vol. 9. — P. 2629-2637.
58. Doshi A.A. A promoter polymorphism of endothelial nitric oxide synthase is associated with reduced mRNA and protein expression in failure human myocardium / A.A. Doshi, M.T. Ziolo // J. Card. Fail. — 2010. — Vol. 16 (4). — P. 314-319.
59. T-786C polymorphism of the NOS-3 gene and the endothelial cell response to fluid shear stress — a proteome analysis / A.R. Asif, M. Oellerich, V.W. Armstrong, M. Hecker, M.J. Cattaruzza // Proteome Res. — 2009. — Vol. 8 (6). — P. 3161-3168.
60. Intracellular processing of endothelial nitric oxide synthase isoforms associated with differences in severity of cardiopulmonary diseases: cleavage of proteins with aspartate vs. glutamate at position 298 / M. Tesauro, W.C. Thompson, P. Rogliani [et al.] // Proc. Natl. Acad. Sci. USA. — 2000. — Vol. 97. — P. 2832-2835.
61. Acidic hydrolysis as a mechanism for the cleavage of the Glu(298)->Asp variant of human endothelial nitric-oxide synthase / T.A. Fairchild, D. Fulton, J.T. Fontana [et al.] // J. Biol. Chem. — 2001. — Vol. 276. — P. 26674-26679.
62. Endotehelial function and carotid intima-media thickness in young healthy subjects among endothelial nitric oxide synthase Glu298Asp and T–>C (-786) polymorphisms / U. Paradossi, J. Ciofini, A. Clerico, N. Botto // Stroke. — 2004. — Vol. 35 (6). — P. 1305-1309.
63. The effects of endothelial nitric oxide synthase gene polymorphisms on endothelial function and metabolic risk factors in healthy subjects: the significance of plasma adiponectin levels / A. Imamura, R. Takahashi, R. Murakami, H. Kataoka, W. Cheng, Y. Numaguchi, T. Murohara, K. Okumura // Eur. J. Endocrinol. — 2008. — Vol. 158 (2). — P. 189-195.
64. Endothelial nitric oxide synthase gene haplotypes affect nitrite levels in black subjects / I.F. Metzger, M.H. Ishizawa, F. Rios-Santos, W.A. Carvalho, J.E. Tanus-Santos // Pharmacogenomics J. — 2011. — Vol. 11 (6). — P. 393-399.
65. Lack of association between endothelial nitric oxide synthase gene polymorphisms, microalbuminuria and endothelial dysfunction in hypertensive men / G. Dell’Omo, G. Penno, L. Pucci, C. Fotino // J. Hypertens. — 2007. — Vol. 25 (7). — Р. 1389-1395.
66. The T->C (-786) and Glu298Asp polymorphisms of the endothelial nitric oxide synthase gene affect th forearm blood flow responses of Caucasian hypertensive patients / G. Rossi, S. Taddei, A. Virdis, M. Cavallin [et al.] // J. Am. Coll. Cardiol.– 2003. — Vol. 41 (6). — Р. 938-945.
67. Biochemical consequences of the NOS3 Glu298Asp variation in human endothelium: altered caveolar localizacion and impaired response to shear / M. Joshi, C. Mineo, P. Shaul, J. Bauer // FASEB J. — 2007. — Vol. 21 (11). — Р. 2655-2663.
68. Полиморфизм гена эндотелиальной NО-синтазы у больных с острыми коронарними синдромами — распространенность, значение для прогноза и выбора тактики лечения / А.Н. Пархоменко, Я.М. Лутай, О.И. Иркин, С.Н. Кожухов, А.А. Скаржевский, А.В. Шумаков, Н.В. Довгань, В.Е. Досенко, А.А. Мойбенко // Український кардіологічний журнал. — 2009. — № 1 (додаток). — С. 15-23.
69. The Glu298Asp polymorphism in the endothelial nitric oxide synthase gene is strongly associated with coronary spasm / K. Chang, S.H. Baek, K.B. Seung, P.J. Kim, S.H. Ihm, J.S. Chae, J.H. Kim // Coron. Aterry Dis. — 2003. — Vol. 14 (4). — P. 293-299.
70. Полиморфизм гена эндотелиальной NO-синтазы и структурно-функциональное состояние крупных сосудов у больных гипертонической болезнью с гипертрофией левого желудочка / О.И. Яковлева, Н.В. Вахрамеева, В.И. Ларионова [и др.] // Артериальная гипертензия. — 2005. — № 3. — С. 195-200.
71. Kose M.I., Akpinar T.S., Bakkaloglu O.K., Tufan A., Sumnu A., Emet S., Kocaaga M., Erk O., Kayacan M.S., Guler K., Demirel A.S. Association of genetic polymorphisms with endothelial dysfunction in chronic heart failure // Eur. Rev. Med. Pharmacol. Sci. — 2014. — Vol. 18 (12). — P. 1755-1761.
72. Поліморфні варіанти Т(-786)С і G894T гена ендотеліальної NO-синтази та стан вазодилатаційної функції ендотелію у хворих із хронічною серцевою недостатністю / Л.Г. Воронков, Н.Г. Горовенко, І.Д. Мазур, І.А. Шкурат, Л.С. Мхітарян, Н.Н. Орлова // Серце і судини. — 2012. — № 4. — С. 43-51.
73. Rossi G.P., Cesari M., Zanchetta M., Colonna S., Maiolino G., Pedon L., Cavallin M., Maiolino P., Pessina A.C. The T-786C endothelial nitric oxide synthase genotype is a novel risk factor for coronary artery disease in Caucasian patients of the GENICA study // J. Am. Coll. Cardiol. — 2003. — Mar 19. — Vol. 41 (6). — P. 930-937.
74. Nishijima T., Nakayama M., Yoshimura M., Abe K., Yamamuro M., Suzuki S., Shono M., Sugiyama S., Saito Y., Miyamoto Y., Nakao K., Yasue H., Ogawa H. The endothelial nitric oxide synthase gene -786T/C polymorphism is a predictive factor for reattacks of coronary spasm // Pharmacogenet Genomics. — 2007. — Aug. — Vol. 17 (8). P. 581-587.
75. Rahimi Z., Nourozi-Rad R. Association of endothelial nitric oxide synthase gene variant (G894T) with coronary artery disease in Western Iran // Angiology. — 2012. — Feb. — Vol. 63 (2). — P. 131-137. doi: 10.1177/0003319711409741. Epub 2011 May 20.
76. Liu D., Jiang Z., Dai L., Zhang X., Yan C., Han Y. Association between the -786T>C 1polymorphism in the promoter region of endothelial nitric oxide synthase (eNOS) and risk of coronary artery disease: a systematic review and meta-analysis // Gene. — 2014. — Jul 15. — Vol. 545 (1). — P. 175-183.
77. Effect of the Asp298 Variant of Endothelial Nitric Oxide Synthase on Survival for Patients With Congestive Heart Failure / M. McNamara Dennis, R. Holubkov, L. Postava, R. Ramani, K. Janosko // Circulation. — 2003. — Vol. 107. — P. 1598-1602.
78. Kim K.M., Murray M.D., Tu W., Robarge J., Ding Y., Brater D.C., Flockhart D.A. Pharmacogenetics and healthcare outcomes in patients with chronic heart failure // Eur. J. Clin. Pharmacol. — 2012. — Nov. — Vol. 68 (11). — P. 1483-1491.
79. Tardin O.M., Pereira S.B., Velloso M.W., Balieiro H.M., Costa B., Alves T.O., Giro C., Pessoa L.P., Ribeiro G.S., Mesquita E.T. Genetic polymorphism G894T and the prognosis of heart failure outpatients // Arq Bras Cardiol. — 2013. — Oct. — Vol. 101 (4). — P. 352-358.
80. Martinelli N.C., Santos K.G., Biolo A., La Porta V.L., Cohen C.R., Silvello D., Andrades M.E., Clausell N., Rohde L.E. Polymorphisms of endothelial nitric oxide synthase gene in systolic heart failure: an haplotype analysis // Nitric Oxide. — 2012. — Mar 31. — Vol. 26 (3). — P. 141-147.
81. Клініко-гемодинамічні показники та довготерміновий клінічний прогноз у пацієнтів із хронічною систолічною серцевою недостатністю залежно від поліморфізму Т(-786)С промотора гена ендотеліальної NO-синтази / Л.Г. Воронков, Н.Г. Горовенко, І.Д. Мазур, І.А. Шкурат, Л.С. Мхітарян, А.В. Ляшенко // Укр. кардіол. журнал. — 2013. — № 1. — С. 55-62.
82. Клініко-гемодинамічні показники та довготерміновий клінічний прогноз у пацієнтів з хронічною систолічною серцевою недостатністю залежно від поліморфізму G894Т гена ендотеліальної NO-синтази / Л.Г. Воронков, Н.Г. Горовенко, І.Д. Мазур, А.В. Ляшенко // Кровообіг і гемостаз. — 2012. — № 3. — С. 55-62.
83. Functional characterization of Glu298Asp mutant human endothelial nitric oxide synthase purified from a yeast expression system / R. Golser, A.C. Gorren, B. Mayer, K. Schmidt // Nitric Oxide. — 2003. — Vol. 8. — P. 7-14.
84. Пархоменко А.Н., Иркин О.И., Лутай Я.М., Степура А.А., Белый Д.А. Эндотелиальная дисфункция у больных с острым инфарктом миокарда: связь с течением заболевания // Укр. кардіол. журнал. — 2013. — № 4 (додаток). — С. 165-166.
Вперше надруковано в журналі «Серцева недостатність», 2015, № 1

/72.jpg)
/74.jpg)
/75.jpg)