Пероксисомные болезни — группа заболеваний, обусловленных нарушением ферментативных реакций внутри органелл, процессов транспорта белков через мембраны органелл и рецепторы [1]. В большинстве случаев патогенетические механизмы пероксисомных болезней связаны с нарушением четырех основных биохимических процессов — синтеза плазмалогенов, окисления жирных кислот с очень длинными цепями и фитановой кислоты, а также дегидратации пипеколиновой кислоты. Традиционно принято выделять три основные формы: гепатоцереброренальный синдром Цельвегера (OMIM214100; МКБ-10: Q87.8), неонатальная Х-сцепленная адренолейкодистрофия (OMIM 202370; МКБ-10: E71.3) и инфантильная болезнь Рефсума (OMIM 266510; МКБ-10: G60.1) [2]. Глобальная фундация пероксисомных заболеваний эту группу заболеваний рекомендует обозначать как пероксисомные биогенные нарушения в спектре синдрома Цельвегера, частота которых составляет 1 : 50 000 новорожденных [3]. Мутации в PEX1 составляют почти 70 % всех случаев PBD-ZSD, 26 % случаев вызваны мутациями в PEX6, PEX10, PEX12 или PEX26 [4, 5].
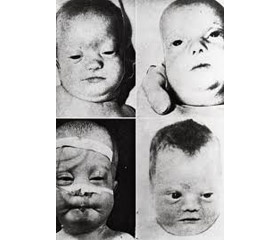
Клинические особенности. Врожденный комплекс черепно-лицевых дисморфий при синдроме Цельвегера считается достаточно специфичным для постановки диагноза по результатам осмотра новорожденного. Он включает: уплощенное лицо и затылок; высокий лоб; гипертелоризм; эпикант; мелкие орбиты; гипоплазию надбровных дуг; высокое, готическое небо; запавшую широкую переносицу; микрогнатию; расхождение швов черепа; увеличение переднего и заднего родничков; низко расположенные уши. Дети с синдромом Цельвегера, наиболее тяжелым синдромом спектра, обычно дебютируют в неонатальном периоде и не выживают до 1 года жизни. Типичными клиническими проявлениями являются гипотония, нарушения питания, снижение слуха и зрения, судороги, что обусловлено распадом миелина нервных волокон и затруднением передачи нервных импульсов (демиелинизацией), приводящим к уменьшению белого вещества (лейкодистрофии). Пациенты с синдромом Цельвегера имеют тяжелые, угрожающие жизни нарушения строения и функции печени, сердца, почек, аномалии скелета, включающие увеличение родничков, наличие характерной зернистости длинных трубчатых костей и позвоночника (chondrodysplasia punctata) [6–10].
Носители генов X-ALD в течение первых нескольких лет жизни имеют бессимптомное течение, после чего появляются признаки недостаточности надпочечников и демиелинизации головного мозга, парапареза, затем дисфункции сфинктера мочевого пузыря. Клиническая картина X-ALD отличается разнообразием проявлений даже в пределах одной семьи. Наиболее частыми формами X-ALD являются церебральная, реже — адренолейкомиелоневропатия, которая развивается на третьем десятилетии жизни. Встречаются также неврологически асимптомные фенотипы X-ALD (20 %) — с недостаточностью надпочечников или без нее. Церебральная форма X-ALD обычно проявляется в детском возрасте и реже встречается у подростков и взрослых. После короткого периода неспецифических симптомов (нарушение внимания и поведения, снижение успеваемости) развивается быстро нарастающая симптоматика (судорожные припадки, атаксия, утрата зрения, нарушение слуха, тетрапарез) и, наконец, апаллический синдром. Средний интервал от первых симптомов до перехода в вегетативное состояние составляет 1,9–2 года. Нередко у больных с X-ALD ошибочно диагностируются подострый склерозирующий панэнцефалит, рассеянный склероз или наследственная параплегия Штрюмпеля. Комбинация жидкостной хроматографии и тандемной масс-спектрометрии (LC-MS/MS) для выявления повышенного уровня жирных кислот с очень длинными цепями в крови рекомендована для диагностики Х-сцепленной адренолейкодистрофии (X-ALD).
Болезнь Рефсума — генетически детерминированное расстройство окисления фитановой кислоты с ее накоплением в тканях организма, приводящим к неврологическим нарушениям, ухудшению зрения, слуха, обоняния, ихтиозным изменениям кожи, нарушениям со стороны сердца. Возраст манифестации симптомов варьирует от 1 до 50 лет. Выделяют взрослую форму заболевания с началом после 10-летнего возраста и инфантильную — у детей 1–10 лет. В первую очередь поражается нервная система. Дегенеративные изменения затрагивают периферические нервные стволы, передние рога и корешки спинного мозга, мозжечковые тракты, зрительные, глазодвигательные, обонятельные и слуховые нервы. Поражается сетчатка глаз с развитием пигментной ретинопатии. Накопление фитановой кислоты в миокарде приводит к формированию кардиомиопатии. Дегенеративные изменения в нервах проводящей системы сердца обусловливают возникновение аритмии. Замещение линолевых и арахидоновых кислот дермы на фитановую кислоту приводит к ихтиозу.
В настоящее время признается возможность диагностики спектра синдрома Цельвегера у пациентов без нарушения зрения и слуха с клиническими признаками периферической нейропатии и/или мозжечковой атаксии [9–10]. В табл. 1 приведены клинические особенности, наблюдаемые в спектре синдрома Цельвегера, в зависимости от тяжести заболевания и возраста. Выраженность и наличие симптомов заболевания имеют прямую зависимость от возраста ребенка.
Хотя частота и соотношение данных признаков описаны в когорте с подмножеством генотипов PEX, распространенность и сроки исходов среди пациентов-детей с синдромом Цельвегера еще недостаточно изучены [11]. Затрудняют диагностику заболевания индивидуальные особенности фенотипа детей с синдромом Цельвегера.
Для лабораторной верификации диагноза используются биохимические, цитохимические, комплементационные и молекулярно-генетические тесты.
Лабораторные критерии. Наиболее значимые биохимические изменения при всех пероксисомных заболеваниях, и в частности при синдроме Цельвегера, включают β-окисление очень длинноцепочечных жирных кислот (жирные кислоты со структурой, включающей более 24 углеродных атомов), а также α-окисление пристановой и фитановой кислот, метаболизма пипеколиновой кислоты и другие пероксисомные дисфункции (табл. 2) [12].
/78-1.jpg)
Спектр Цельвегера может быть диагностирован при выявлении различных изменений биохимических функций, которые могут быть выявлены в крови и моче (рис. 1). Первый шаг в диагностике пероксисомных болезней включает определение уровня VLCFA в плазме натощак [13]. Рекомендовано определение С26:0 и С26:1 жирных кислот и соотношений С24:0/С22:0, а также С26:0/С22:0, повышение которых является индикатором дефекта в β-окислении жирных кислот с очень длинными цепями [12]. Отметим, что в течение дня количество и соотношение жирных кислот с очень длинными цепями изменяются рандомно, что определяет сомнительные результаты. В случае повышения С26:0 с нормальным соотношением С24:0/С22:0, С26:0/С22:0 и высоким уровнем жирных кислот в крови рекомендовано повторение исследования натощак, после ночного сна. Ложноположительные результаты редки, но возможны у пациентов, находящихся на кетогенной диете.
/79-1.jpg)
Дополнительные исследования обычно демонстрируют повышение уровня пристановой и фитановой кислот, пипеколиновой кислоты и промежуточных метаболитов желчных кислот в крови и моче [14, 15]. Недостаток плазмалогенов в эритроцитах, биосинтез которых связан с пероксисомной функцией, может быть исследован в зависимости от тяжести заболевания. Следует отметить, что у детей в периоде новорожденности уровень пипеколиновой кислоты рекомендовано исследовать в моче, а в более старшем возрасте — в крови [16–18]. Фитановая и пристановая кислоты обычно не повышены у детей грудного возраста, так как они не потребляют продукты, богатые этими кислотами. Кроме того, отложение в печени С27 желчной кислоты, накопление интермедиатов ди- и тригидро–ксихолестеновой кислоты в плазме и моче являются индикатором дефекта их биосинтеза в пероксисомах печени.
Биохимические исследования фибробластов наиболее часто включают анализ окисления фитановой и пристановой кислот, аккумуляции и окисления жирных кислот с очень длинными цепями. Культуры фибробластов кожи демонстрируют дисфункцию пероксисомальных матриксных протеинов внутриклеточной локализации, а именно недостаток пероксисомной каталазы и повышение цитозольной каталазы. Нарушение соотношения внутриклеточных каталаз является индикатором как пероксисомных болезней в спектре синдрома Цельвегера, так и выборочных дефектов ферментов [13, 19]. Около 10–15 % детей с повышением уровня VLCFA в крови имеют не пероксисомные болезни спектра синдрома Цельвегера, а отдельные дефекты β-окисления энзимов в очень длинной цепи –acyl-CoA оксидазы (АСОХ1) и D-бифункционального протеина (HSD17B4). Клинический фенотип этих пациентов относится к атипичным. Другие фенотипы (болезнь Рефсума) включают дефект одного фермента/протеина в жирных кислотах с разветвленной цепью, а также в желчной кислоте, включая α-метил-ацил-СоА рацемазу (AMACR), фитаноил-коэнзим А гидроксилазу (PHYH), PEX7 и носителя протеина Х (SCPx). Так, диагностика пероксисомных заболеваний спектра синдрома Цельвегера не может быть основана только на скрининге уровня VLCFA в крови у пациентов с явными клиническими признаками заболевания. В небольшом числе наблюдений мутации в генах PEX, таких как PEX2, PEX10, PEX12, PEX16, PEX11В, могут быть выявлены у пациентов с отсутствием повышения уровня VLCFA в крови. Следовательно, проведение нескольких биохимических тестов крови, исследование фибробластов кожи и генетическое тестирование необходимы для установки правильного диагноза [20–25].
Генетическое тестирование обеспечивается с помощью методов секвенирования нового поколения для генов PEX. Техника секвенирования нового поколения — определение нуклеотидной последовательности ДНК и РНК для описания первичной структуры позволяет «прочитать» единовременно сразу несколько участков генома. Пациенты с двумя нулевыми аллелями PEX обычно имеют тяжелое течение заболевания, и, напротив, у детей с –
PEX1-p.G843D-генотипом заболевание будет протекать легче. Гомозиготы с PEX1-p.G843D имеют наиболее легкое течение заболевания, однако в фенотипе присутствуют нарушения интеллекта. Комбинация нулевого аллеля PEX с миссенс-мутацией (точечная мутация, в результате которой измененный кодон начинает кодировать другую аминокислоту) может варьироваться от среднего до более легкого течения заболевания и зависит от остаточной миссенс-мутации. В недавних публикациях описаны миссенс-мутации в PEX1 и PEX6 в сочетании с нулевыми аллелями у детей с пероксисомными болезнями спектра синдрома Цельвегера и нормальным интеллектом. Кроме того, у пациентов с мутациями в области, кодирующей «цинковый палец» PEX2, PEX12 и PEX10, и с некоторыми мутациями в PEX16 отмечаются различные варианты фенотипов. В отличие от биохимических тестов мутационный анализ также идентифицирует гетерозиготные носители, которые могут обеспечить надежное генетическое консультирование семей с риском развития пероксисомных заболеваний [26–28].
Неонатальный скрининг. Комбинация жидкостной хроматографии и тандемной масс-спектрометрии (LC-MS/MS) для выявления повышенных уровней VLCFA у новорожденных в пятнах крови была подтверждена как скрининговый метод для Х-сцепленной адренолейкодистрофии (X-ALD), связанного с ним пероксисомального расстройства [6, 41, 42]. В настоящее время скрининг новорожденных проводится в некоторых штатах США (Нью-Джерси, Коннектикут, Иллинойс, Теннесси и Калифорния) и в Нью-Йорке, что позволяет в ранние сроки обеспечить менеджмент пациентов. В будущем в США планируется расширение спектра скрининговой диагностики пероксисомных заболеваний у новорожденных [29–30].
Пренатальная диагностика континиума синдрома Цельвегера выполняется в первом или во втором триместре беременности с использованием биохимического или генетического тестирования хорионических ворсинок клеток или культивируемых амниоцитов. Возможна предимплантационная генетическая диагностика, когда известны мутации гена PEX [31].
Мониторинг и лечение. Пероксисомные заболевания спектра синдрома Цельвегера относятся к полиорганным заболеваниям с функциональными нарушениями пероксисом, участвующих в метаболических процессах всех клеток организма начиная с пренатального периода.
Клинический мониторинг с момента постановки диагноза включает коррекцию исходных нарушений и выявления симптомов заболевания, которые появляются позднее (табл. 3) [49].
/80-1.jpg)
Диспансеризация детей обычно проводится ежегодно (в отдельных случаях — чаще). Аномалии развития головного мозга, почек, скелета, выявленные в неонатальном периоде, демонстрируют тяжелое течение спектра. Мониторинг таких пациентов более предсказуемый, чем при легких формах. Дети с тяжелым течением имеют краниофасциальный дисморфизм, а именно увеличение лицевой части черепа, выдающийся лоб, широкую и уплощенную переносицу, а также микрогнатию. У пациентов с тяжелым течением, по данным магнитно-резонансной томографии, наблюдаются неонатальные судороги, тяжелая гипотония, стагнация развития, микрогирия и гетеротопия серого вещества головного мозга. Судороги часто резистентны к терапии противоэпилептическими средствами. Возможны мальформации внутренних органов: ренальные кисты, типично увеличение печени с дисфункцией гепатоцеллюлярной и билиарной систем. В связи с диффузной гипотонией у детей часто наблюдаются нарушение глотания, дефицит массы тела, что вынуждает к установке гастрального зонда. Также характерны ларингомаляция и другие респираторные дисфункции, требующие использования назальных канюль для оксигенации. Применение более агрессивных технологий респираторной поддержки обычно обсуждается с родителями с учетом прогнозов для выживаемости и качества жизни. Психофизическое развитие ребенка обычно недостаточное или вовсе наблюдается стагнация [32, 33].
Для большинства пациентов с легкой и средней степенями тяжести синдрома Цельвегера мониторинг и менеджмент описаны ниже.
Кормление и нутритивная поддержка. Многие дети с PBD-ZSD к пище относятся избирательно, что определяет их участие в программах поведенческого питания. В некоторых случаях необходима установка гастрального зонда или гастростомы. В питании преобладают легкоусвояемые продукты с учетом дефицита желчных кислот, ассоциированного с мальабсорбцией. В настоящее время диета, рекомендуемая детям с синдромом Цельвегера, не разработана. Хотя уровни VLCFA повышены в тканях и пациентов с данным заболеванием, пока не выяснено, предотвратит ли снижение VLCFA в диете прогрессирование заболевания или связанные с ним симптомы [34]. На сегодняшний день корреляции между снижением количества потребляемого VLCFA и содержания его в крови не выявлено, поскольку большинство VLCFA организм вырабатывает эндогенно. Уровни плазменной VLCFA снижаются только комбинацией диетического восстановления VLCFA и добавления масла Лоренцо (смесь 4 : 1 триолеата глицерина и триэфирата глицерина) у пациентов с X-ALD [35–37], но это не влияет на прогрессирование уже установленной лейкодистрофии. Более того, диета с повышенным содержанием мононенасыщенных жирных кислот в масле Лоренцо может быть противопоказана у пациентов с собственно синдромом Цельвегера, которые накапливают большое количество C26:1 из-за дефектного окисления VLCFA [38, 49].
Фитановая кислота представляет собой исключительно жирную кислоту с разветвленной цепью. Она находится в жирах жвачных животных, молочных продуктах и некоторых сортах рыбы [39]. Таким образом, повышение уровня фитановой кислоты в плазме можно устранить с помощью диетического ограничения [39]. Существует незначительное количество фитановой кислоты в грудном молоке человека [38]. Диетическое ограничение фитановой кислоты рекомендовано детям, особенно с болезнью Рефсума, однако неэффективно при нормальном уровне фитановой кислоты в плазме, поскольку у пациентов нарушается эндогенный синтез докозагексаеновой кислоты (ДГК) (незаменимая полиненасыщенная жирная кислота класса омега-3), а она важна для развития мозга и сетчатки. Добавление ДГК в пищу было ранее рекомендовано. Однако плацебо-контролируемое исследование, проведенное в 2010 году, показало, что клинической пользы от приема ДГК не было [40].
Рекомендуется добавление в пищу жирорастворимых витаминов, A, D, E и K, поддержка гепатобилиарной системы [41]. Для поддержания функции печени рекомендована дотация витамина К 2,5–5 мг в сутки. Так как метаболизм желчных кислот нарушен, желательна терапия желчными кислотами (холевая кислота и хенодезоксихолевая кислота). Холевая кислота одобрена управлением по контролю за продуктами и лекарствами США для лечения пероксисомальных расстройств, включая спектр Цельвегера [41]. Лица с дисфункцией печени требуют более частого мониторинга и наблюдения гастроэнтеролога, редко — хирурга и гематолога (дисфункция печени может способствовать варикозу вен пищевода).
Коррекция слуха. У многих пациентов с PBD-ZSD наблюдается некоторое снижение слуха [42], поэтому данная функция оценивается. Для коррекции слуха используют слуховые аппараты и кохлеарные имплантаты, что улучшает восприятие и в некоторых случаях речь ребенка [43].
Коррекция зрения. В результате дистрофии сетчатки и аномалии зрительного нерва, катаракты (редко) у детей с синдромом Цельвегера наблюдаются снижение зрения и слепота [44, 45], что определяет ежегодный мониторинг зрения. Очки рекомендованы для коррекции рефракции. Пока не доказана эффективность электроретинограммы для оценки функции зрения, выполнение оптической когерентной томографии у детей, которые могут быть в комплайенсе с исследователем, глядя прямо на источник света, полезно для определения и мониторинга состояния сетчатки.
Лечение неврологических нарушений. Судороги наблюдаются почти у всех пациентов с тяжелым течением и у 23 % детей с более легкими формами заболевания. Электроэнцефалография (ЭЭГ) выполняется каждый раз, когда происходят изменения в неврологическом статусе ребенка. У пациентов со спектром Цельвегера может формироваться лейкодистрофия [46], поэтому рекомендована МРТ. Для лечения судорог рекомендовано использование леветирацетама, фенобарбитала, клоназепама, топирамата и ламотриджина.
Мониторинг костной ткани и лечение ее деминерализации. Дети с тяжелым течением заболевания могут иметь зернистость длинных трубчатых костей и позвоночника или штрихование в зонах роста. Снижение минерализации кости, которое ухудшается с течением времени, ассоциировано с легкой и средней тяжести формами заболевания. У детей с задержкой роста и переломами в анамнезе рекомендованы проведение двухэнергетической рентгеновской абсорбциометрии, определение уровня витамина D и фосфора в крови, а также паратиреоидного гормона [47]. Недавние исследования показали успешность лечения бисфосфонатными препаратами болезни костей в спектре синдрома Цельвегера [47], однако данное заключение требует обсуждения. В то же время показано, что физическая нагрузка уменьшает скорость деминерализации костей и способствует росту детей с данным спектром. Коррекция аномалий зубной эмали проводится стоматологом. Стоматологическое обследование должно осуществляться детям каждые 6 месяцев.
Лечение недостаточности надпочечников. Первичная надпочечниковая недостаточность характерна для синдрома Цельвегера, особенно для X-сцепленной адренолейкодистрофии [47]. Рекомендуется ежегодный (или более частый) мониторинг адренокортикотропного гормона и утреннего кортизола. Лечение включает стандартную заместительную терапию.
У детей старшего возраста (≥ 4–6 лет) следует контролировать гипероксалурию. Мониторируют уровень щавелевой кислоты и креатинина в моче. УЗИ почек может проводиться с целью выявления микро- и макролитов.
Также рекомендуется, чтобы все пациенты в спектре синдрома Цельвегера были вакцинированы против гриппа и респираторно-синцитиального вируса ежегодно.
Ниже приводим собственное наблюдение диагностики и особенностей клинической картины у ребенка грудного возраста, которые дали основание заподозрить редкое пероксисомное заболевание спектра Цельвегера (X-ALD). Тяжесть течения заболевания, скорость развития полиорганной недостаточности не позволили обследовать ребенка в полном объеме, однако представляем клинические особенности и некоторые биохимические признаки заболевания.
Ребенок К., 2 месяца 19 дней, мальчик. Поступил впервые в тяжелом состоянии с жалобами на плохую прибавку массы тела, срыгивания и рвоту «фонтаном», задержку темпов психомоторного развития, беспокойство, субфебрильную температуру тела.
Из анамнеза жизни и заболевания известно, что ребенок от второй беременности (1-я беременность — девочка 2 лет, диагноз: фенилкетонурия), протекавшей на фоне острой респираторной инфекции, от 2-х срочных родов, была пренатально низко расположена плацента (по данным УЗИ). Масса тела при рождении — 2600 г, длина — 50 см. С 2 недель жизни искусственное вскармливание (коррекция питания в виде замены смесей проводилась 9 раз). Обследован в многопрофильном стационаре г. Одессы (проводилась дифференциальная диагностика между врожденной дисфункцией коры надпочечников и нарушением обмена веществ). С целью уточнения диагноза, подбора терапии, коррекции питания госпитализирован в ОДКБ. По материнской линии: у тети и дяди ребенка рассеянный склероз, двоюродная сестра имеет трисомию по 21-й хромосоме. У прабабушки двое детей умерли в возрасте 2 и 4 недели, 1 ребенок родился мертворожденным.
Анамнез заболевания: болел с рождения (имели место плохая прибавка в массе, снижение толерантности к пище, вялость). В возрасте одного месяца диагностированы белково-энергетическая недостаточность, гипоксически-ишемическая энцефалопатия, синдром двигательных нарушений. При обследовании выявлены повышение аспарагиновой и аланиновой аминотрансфераз, киста сосудистого сплетения и неоднородность эхоструктуры левого надпочечника (по данным УЗИ). Проведена коррекция водно-электролитного баланса, белково-энергетической недостаточности, что улучшило состояние ребенка. Спустя две недели появилась рвота «фонтаном», беспокойство. С диагнозом «врожденная дисфункция коры надпочечников» ребенок направлен в педиатрическое отделение для детей с множественными аномалиями развития и редкими (орфанными) заболеваниями. В течение 13 дней находился в отделении редких заболеваний. В динамике нарастали вялость, адинамия, угасал сосательный рефлекс, появились геморрагические вкрапления в рвотных массах (желудочно-кишечное кровотечение исключено), в связи с чем ребенок переведен в отделение ОАиИТ. На фоне проводимой терапии состояние ребенка прогрессивно ухудшалось за счет проявлений церебральной, дыхательной, сердечно-сосудистой недостаточности, коагулопатии. В течение 14 часов ребенок находился на искусственной вентиляции легких. Обращали на себя внимание фенотипические особенности: сплюснутое лицо, увеличенный большой родничок, короткий вздернутый нос, широкая переносица, высокий лоб, уплощенный затылок, монголоидный разрез глаз, эпикантальные складки, избыточные кожные складки на шее. Отмечались стагнация физического развития, гипотония, гипотрофия, гепатомегалия.
Дополнительные лабораторные исследования:
— биохимическое исследование крови: ↓ железа — 5 (7,2–17,9), ↓ холестерина — 2,42 (2,95–5,25), ↓ глюкозы — 2,91 (3,3–5,6), ↓ мочевой кислоты — 0,31 (1,38–2,98), ↓ Р — 1,44 (1,45–2,16), ↓ белка — 40,66 (60–78), ↓ альбумина — 36,16 (38–54); ↑ ЩФ — 1184,6 (до 1107), ↑ билирубина — 29,86 (до 20,5), ↑ ГГТ — 738,25 (до 204); уровень АСТ, АЛТ, триглицеридов, мочевины, магния, креатинина, креатинфосфокиназы, лактатдегидрогеназы, амилазы не изменен;
— исследованы маркеры гепатита В и С — отрицательные;
— исследование эндокринной системы (анализ крови тиреоидной группы, гормоны надпочечников, гипоталамуса и гипофиза): уровень тиреотропного гормона, кортизола, адренокортикотропного гормона в пределах нормальных значений; ↑ 17-ОН прогестерона крови — 18,0 (норма — 0,1–2,7), ↑ калия крови — 5,72 (3,5–5,3), ↓ натрия крови — 131,6 (135–148);
— анализ мочи методом газовой хроматографии и масс-спектрометрии — признаки лактат-ацидоза, выявлены изменения метаболитов жирных кислот, признаки дефицита В2, В3, В5;
— высокоэффективная жидкостная хроматография аминокислот — ↑ метионина, цистина, фенилаланина, тирозина, глутамина, цитруллина, аспарагина, аргинина, орнитина, аланина, лейцина, изолейцина, валина, треонина, пролина, глицина, серина; ↓ триптофана, аспартата;
— кариотипирование крови (анализ 15 метафаз культуры лимфоцитов периферической крови) показало нормальный мужской кариотип с увеличением гетерохроматина в длинном плече Y-хромосомы (полиморфизм — вариант нормы согласно ISCN 2013 г.);
— полиморфные варианты генов ферментов фолатного цикла: MTHFR C677Т, MTRR A66G, MTR A2756G, факторы свертывания: F II G20120A,
F V R506Q без патологии;
— тесты на полиморфизм C13910T (LCT) — выявлены аллели Т/Т (генотип связан с нормальной переносимостью лактозы).
Дополнительные инструментальные исследования:
— ультразвуковое исследование сердца: врожденный порок сердца (двухстворчатый аортальный клапан);
— ультразвуковое исследование почек: ретенционные кисты почек;
— ультразвуковое исследование надпочечников: неоднородность эхоструктуры левого надпочечника (что подтверждено результатами посмертного макроскопического исследования — «гофрированные» надпочечники);
— магнитно-резонансная томография головного мозга: пери- и суправентрикулярно определяются гиперинтенсивные в Т2 и tirm зоны, незаращение передних отделов прозрачной перегородки. Конвекситальные ликворосодержащие пространства расширены в лобно-височно-теменных отделах до 4–5 мм. Отмечается высокое стояние большой цистерны мозга.
Клинические, лабораторные и инструментальные данные дали основания предположить наличие у ребенка одного из пероксисомных заболеваний спектра Цельвегера, что подтверждено повышением уровня VLCFA в крови (метаболиты окисления жирных кислот: ↑↑↑ 3-hydroxybutyric, ↑ acetoacetatic, ↑ suberic, ↑↑↑ 3-hydroxysebacic). Генетическое тестирование мутации ABCD1 гена, последовательность ДНК в гене PEX и связанные с ними мутации генов отдельных пероксисомных ферментов провести не удалось ввиду прогредиентного течения заболевания, полиорганной недостаточности, которые привели к летальному исходу в возрасте 2 месяца 28 дней. Результаты ауто–псии подтвердили вероятность пероксисомного заболевания спектра синдрома Цельвегера.
Таким образом, представленные принципы диагностики и лечения пероксисомных заболеваний спектра синдрома Цельвегера, пример возможностей диагностики заболевания в Украине представляют собой отправную точку медицинских мероприятий. Эти принципы будут развиваться со временем, поскольку исследуются новые терапевтические стратегии. На моделях человеческого организма in vitro проводится культивирование клеток пациентов, включая фибробласты кожи, что может обеспечить эффективный скрининг и лекарственную терапию in vitro. Недавно фибробласты кожи, полученные у ребенка с синдромом Цельвегера, были перепрограммированы в индуцированные плюрипотентные стволовые клетки (iPSC), которые дифференцированы в нейронные и печеночные модели клеток. Существует несколько генетически модифицированных моделей мышиных дефектов гена PEX, включая модель общей мутации
PEX1 p.G843D. Имеются биохимические банки веществ, таких как бетаин, аргинин, которые in vitro улучшают функцию пероксисом. Признаются потенциальные терапевтические возможности для клеточной терапии, включая трансплантацию типов клеток и клеточных линий, затронутых у пациентов с синдромом Цельвегера [47].
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Braverman N.E. Peroxisome biogenesis disorders: Biological, clinical and pathophysiological perspectives / N.E. Braverman, M.D. D’Agostino, G.E. Maclean // Dev. Disabil. Res. Rev. — 2013. — № 17(3). — P. 187-196.
2. Zellweger H. History of the cerebrohepatorenal syndrome of Zellweger and other peroxisomal disorders / H. Zellweger, P. Maertens, D. Superneau, W. Wertelecki // South. Med. J. — 1988. — № 81(3). — P. 357-364.
3. Steinberg S.J. Peroxisome biogenesis disorders / S.J. Steinberg, G. Dodt, G.V. Raymond, N.E. Braverman, A.B. Moser, H.W. Mo–ser // Biochim. Biophys. Acta. — 2006. — № 1763(12). — P. 1733-1748.
4. Yik Y. Identification of novel mutations and sequence variation in the Zellweger syndrome spectrum of peroxisome biogenesis disorders / Y. Yik, S.J. Steinberg, A.B. Moser et al. // Hum. Mutat 2009. — № 30(3). — P. 467-480.
5. Ebberink M.S. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder / M.S. Ebberink, P.A. Mooijer, J. Gootjes, J. Koster et al. // Hum. Mutat. — 2011. — № 32(1). — P. 59-69.
6. Ebberink M.S. Identification of an unusual variant peroxisome biogenesis disorder caused by mutations in the PEX16 gene / M.S. Ebberink, B. Csanyi, W.K. Chong et al. // J. Med. Genet. — 2010. — № 47(9). — P. 608-615.
7. Regal L. Mutations in PEX10 are a cause of autosomal recessive ataxia / L. Regal, M.S. Ebberink, N. Goemans et al. // Ann. Neurol. — 2010. — № 68(2). — P. 259-263.
8. Sevin C. Autosomal recessive cerebellar ataxia caused by mutations in the PEX2 gene / C. Sevin, S. Ferdinandusse, H.R. Waterham et al. // Orphanet J. Rare Dis. — 2011. — № 6.
9. Majewski J. A new ocular phenotype associated with an unexpected but known systemic disorder and mutation: novel use of genomic diagnostics and exome sequencing / J. Majewski, Z. Wang, I. Lopez et al. // J. Med. Genet. — 2011. — № 48(9). — P. 593-596.
10. Ratbi I. Heimler syndrome is caused by hypomorphic mutations in the peroxisomebiogenesis genes PEX1 and PEX6 / I. Ratbi, K.D. Falkenberg, M. Sommen et al. // Am. J. Hum. Genet. — 2015. — № 97(4). — P. 535-545.
11. Poll-The B.T. Peroxisome biogenesis disorders with prolonged survival: phenotypic expression in a cohort of 31 patients / B.T. Poll-The, J. Gootjes, M. Duran et al. // Am. J. Med. Genet. — 2004. — № 126(4). — P. 333-338
12. Wanders R.J. Biochemistry of mammalian peroxisomes revisited / R.J. Wanders, H.R. Waterham // Annu. Rev. Biochem. — 2006. — № 75. — P. 295-332.
13. Steinberg S. Investigational methods for peroxisomal disorders / S. Steinberg, R. Jones, C. Tiffany, A. Moser // Curr. Protoc. Hum. Genet. — 2008. — № 97(4). — P. 535-545.
14. Stradomska T.J. The impact of a ketogenic diet and liver dysfunction on serum very long-chain fatty acids levels / T.J. Stradomska, M. Bachanski, J. Pawlowska et al. // Lipids. — 2013. — № 48(4). — P. 405-409.
15. Theda C. Increased very long chain fatty acids in patients on a ketogenic diet: a cause of diagnostic confusion / C. Theda, R.C. Woody, S. Naidu et al. // Molecular Genetics and Metabolism. — 2016. — № 117. — P. 313-321.
16. Peduto A. Hyperpipecolic acidaemia: a diagnostic tool for pe–roxisomal disorders / A. Peduto, M.R. Baumgartner, N.M. Verhoeven et al. // Mol. Genet. Metab. — 2004. — № 82(3). — P. 224-230.
17. Waterham H.R. Genetics and molecular basis of human pe–roxisome biogenesis disorders / H.R. Waterham, M.S. Ebberink // Biochim. Biophys. Acta. — 2012. — № 1822 (9). — P. 1430-1441.
18. Steinberg S.J. A PEX10 defect in a patient with no detectable defect in peroxisome assembly or metabolism in cultured fibroblasts / S.J. Steinberg, A. Snowden, N.E. Braverman et al. // J. Inherit. Metab. Dis. — 2009. — № 32(1). — P. 109-119.
19. Krause C. Rational diagnostic strategy for Zellweger syndrome spectrum patients / C. Krause, H. Rosewich, J. Gartner // Eur. J. Hum. Genet. — 2009. — № 17(6). — P. 741-748.
20. Ferdinandusse S. Clinical, biochemical, and mutational spectrum of peroxisomal acyl-coenzyme A oxidase deficiency / S. Ferdinandusse, S. Denis, E.M. Hogenhout et al. // Hum. Mutat. — 2007. — № 28(9). — P. 904-912.
21. Lieber D.S. Next generation sequencing with copy number variant detection expands the phenotypic spectrum of HSD17B4-deficiency / D.S. Lieber, S.G. Hershman, N.G. Slate et al. // BMC Med. Genet. — 2014. — № 15. — P. 30.
22. Haugarvoll K. MRI characterisation of adult onset alphamethylacyl-coA racemase deficiency diagnosed by exome sequencing / K. Haugarvoll, S. Johansson, C. Tzoulis et al. // Orphanet J. Rare Dis. — 2013. — № 8. — P. 1.
23. Wanders R.J.A. Refsum disease / R.J.A. Wanders, H.R. Waterham, B.P. Leroy // GeneReviews. — 2006. — № 20. — P. 1993-2015.
24. Wierzbicki А.S. Peroxisomal disorders affecting phytanic acid alpha-oxidation: a review / А.S.Wierzbicki // Biochem. Soc. Trans. — 2007. — № 35. — P. 881-886.
25. Ferdinandusse S. Mutations in the gene encoding peroxisomal sterol carrier protein X (SCPx) cause leukencephalopathy with dystonia and motor neuropathy / S. Ferdinandusse, P. Kostopoulos, S. Denis et al. // Am. J. Hum. Genet. — 2006. — № 78 (6). — P. 1046-1052.
26. Levesque S. A founder mutation in the PEX6 gene is responsible for increased incidence of Zellweger syndrome in a French Canadian population / S. Levesque, C. Morin, S.P. Guay, J. Villeneuve et al. // BMC Med. Genet. — 2012. — № 13. — P. 72.
27. Sun Y. Analysis of a Chinese pedigree with Zellweger syndrome reveals a novel PEX1 mutation by next-generation sequen–cing / Y. Sun, L. Wang, X. Wei, Q. Zhu et al. // Clin. Chim. Acta. — 2013. — № 417. — P. 57-61.
28. Buchert R.H. A peroxisomal disorder of severe intellectual disability, epilepsy, and cataracts due to fatty acyl-CoA reductase 1 deficiency / R. Buchert, H. Tawamie, C. Smith et al. // Am. J. Hum. Genet. — 2014. — № 95 (5). — P. 602-610.
29. Hubbard W.C. Newborn screening for X-linked adrenoleukodystrophy (X-ALD): validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method / W.C. Hubbard, A.B. Moser, A.C. Liu, R.O. Jones, S.J. Steinberg, F. Lorey, S.R. Panny, R.F. Vogt Jr., D. Macaya, C.T. Turgeon et al. // Mol. Genet. Metab. — 2009. — № 97(3). — P. 212-220.
30. Vogel B.H. Newborn screening for X-linked adrenoleukodystrophy in New York State: diagnostic protocol, surveillance protocol and treatment guidelines / B.H. Vogel, S.E. Bradley, D.J. Adams et al. // Mol. Genet. Metab. — 2015. — № 114(4). — P. 599-603.
31. Al-Sayed M. Preimplantation genetic diagnosis for Zellweger syndrome / M. Al-Sayed, S. Al-Hassan, M. Rashed, M. Qeba, S. Coskun // Fertil. Steril. — 2007. — № 87(6). — P. 1468.
32. Lee P.R. Child neurology: Zellweger syndrome / P.R. Lee, G.V. Raymond // Neurology. — 2013. — № 80(20). — P. 207-210.
33. Barkovich A.J. MR of Zellweger syndrome / A.J. Barkovich, W.W. Peck // AJNR Am. J. Neuroradiol. — 1997. — № 18(6). — P. 1163-1170.
34. Van Duyn M.A. The design of a diet restricted in saturated very long-chain fatty acids: therapeutic application in adrenoleukodystrophy / M.A. Van Duyn, A.E. Moser, F.R. Brown et al. // Am. J. Clin. Nutr. — 1984. — № 40(2). — P. 277-284.
35. Aubourg P. A two-year trial of oleic and erucic acids (“Lorenzo’s oil”) as treatment for adrenomyeloneuropathy / P. Aubourg, C. Adamsbaum, M.C. Lavallard-Rousseau et al. // N. Engl. J. Med. — 1993. — № 329(11). — P. 745-752.
36. Rizzo W.B. Dietary erucic acid therapy for X-linked adrenoleukodystrophy / W.B. Rizzo, R.T. Leshner, A. Odone et al. // Neurology. — 1989. — № 39 (11). — P. 1415-1422.
37. Uziel G. Experience on therapy of adrenoleukodystrophy and adrenomyeloneuropathy / G. Uziel, E. Bertini, P. Bardelli et al. // Dev. Neurosci. — 1991. — № 13(4–5). — P. 274-279.
38. Egge H. Minor constituents of human milk (I) identification of cyclohexaneundecanoic acid and phytanic acid in human milk fat by a combination gas chromatograph-mass spectrometer / H. Egge, U. Murawski, P. Gyorgy et al. // FEBS Lett. — 1969. — № 2(4). — P. 255-258.
39. Watkins P.A. Identification of differences in human and great ape phytanic acid metabolism that could influence gene expression profiles and physiological functions / P.A. Watkins, A.B. Moser, C.B. Toomer et al. // BMC Physiol. — 2010. — № 10. — P. 19.
40. Paker A.M. Docosahexaenoic acid therapy in peroxisomal diseases: results of a double-blind, randomized trial / A.M. Pa–ker, J.S. Sunness, N.H. Brereton et al. // Neurology. — 2010. — № 75(9). — P. 826-830.
41. Clayton P.T. Inborn errors of bile acid metabolism / P.T. Clayton // J. Inherit. Metab. Dis. — 1991. — № 14(4). — P. 478-496.
42. Theil A.C. Clinical recognition of patients affected by a pe–roxisomal disorder: a retrospective study in 40 patients / A.C. Theil, R.B. Schutgens, R.J. Wanders, H.S. Heymans // Eur. J. Pediatr. — 1992. — № 151(2). — P. 117-120.
43. Broomfield S.J. Cochlear implantation in children with syndromic deafness / S.J. Broomfield, I.A. Bruce, L. Henderson et al. // Int. J. Pediatr. Otorhinolaryngol. — 2013. — № 77(8). — P. 1312-1316.
44. Wilson G.N. Zellweger syndrome: diagnostic assays, syndrome delineation, and potential therapy / G.N. Wilson, R.G. Holmes, J. Custer et al. // Am. J. Med. Genet. — 1986. — № 24(1). — P. 69-82.
45. Hittner H.M. Zellweger syndrome. Lenticular opacities indicating carrier status and lens abnormalities characteristic of homozygotes / H.M. Hittner, F.L. Kretzer, R.S. Mehta // Arch. Ophthalmol. — 1981. — № 99(11). — P. 1977-1982.
46. Maeda K. Oral bile Acid treatment in two Japanese patients with Zellweger syndrome / K.Maeda, A. Kimura, Y. Yamato et al. // J. Pediatr. Gastroenterol. Nutr. — 2002. — № 35(2). — P. 227-230.
47. Behringer M. Effects of weight-bearing activities on bone mine–ral content and density in children and adolescents: a meta-analysis / M. Behringer, S. Gruetzner, M. McCourt, J. Mester // J. Bone Miner. Res. — 2014. — № 29(2). — P. 467-478.

/77-1.jpg)